Federico
Soriguer.
Médico
endocrinólogo. Miembro de la Academia Malagueña de Ciencias
(Conferencia pronunciada en la clausura de las III Jornadas sobre infancia y adolescencia)


Introducción
Que el hombre es un continuo con la naturaleza ha formado
parte de algunas corrientes filosóficas, como la de los epicúreos, pero más
como una intuición que como un hecho. En, Sobre la
naturaleza de las cosas. ( en De rerum natura de
Tito Lucrecio Caro, un poema compuesto en torno al año 50 antes de Cristo,
podemos leer: “ todo
está hecho de partículas invisibles (los átomos); el universo, no tiene creador
ni propósito, no fue creado para los humanos, que no son seres únicos; la
sociedad no comenzó en una supuesta edad de oro sino en una lucha por la
supervivencia; no existe el más allá; las religiones organizadas son fruto de
la superstición y se sostienen por la crueldad y el miedo; el fin de la vida
humana es la reducción del dolor y la búsqueda del placer”.
Pero no ha sido hasta hace un
par de siglos cuando los humanos tomamos conciencia clara de que:
1. Somos
un continuo evolutivo que comenzó hace, probablemente 4 mil millones de año, o
quizás más, a partir de una forma
primordial de vida, que se ha ido organizando gracias al azar y a la necesidad
(Monot) complejizándose y especializándose en función de los mecanismos que
desde Darwin conocemos como teoría de la
evolución.
2. Nuestro
cuerpo es el resultado de un programa que aparece en el momento mismo de la
formación del huevo a partir a partir
de dos gametos (masculino y femenino) que portan ya toda la información,
incluido la de un reloj biológico que comienza
en el momento mismo de la fecundación y que va desarrollando y
condicionando la embriogénesis (durante el embarazo), el nacimiento,
crecimiento, maduración sexual, la vida adulta, el envejecimiento y la muerte.
3. Y,
finalmente, que la cultura, (cualquiera que sea la forma en la que la
entendamos, (ver más adelante), en fin,
es parte de la propia naturaleza humana, pues procede de ella, de la capacidad
tecnológica que define al propio ser como humano y que a su vez condiciona e
influye en ese reloj o programa biológico.
Se suele llamar filogenia
al primer punto, ontogenia al
segundo y al tercero, simplemente cultura
(Fig. 1).
Hoy es difícil comprender la naturaleza humana sin tener
en cuenta las estrechas relaciones entre la filogenia, la ontogenia y la
cultura. En este capítulo de aborda la cuestión de la pubertad y la
adolescencia desde estas coordenadas bajo la influencia de los trabajos más
recientes de la biología evolutiva a través de los modelos lalmados EVO-DEVO,
que, de alguna manera, recuperan las
tesis de Philip August Haeckel (1834-1919) resumida en la famosa frase : la
Ontogenia recapitula de Filogenia.
Fig.
1. Filogenia, Ontogenia, Cultura. Bases de un modelo EVO-DEVO

Pubertad
y adolescencia, definición:
El
legado de Tanner
JM Tanner publicó en 1955 y revisó en 1962, “Growth
y Adolescense”[1] . en el que planteaba
hipótesis y propuestas sobre la adolescencia y la pubertad, que siguen
vigentes, como la disminución secular de la edad de la menarquía, la
comparación de la pubertad humana con la de algunos primates o la sincronía entre el
crecimiento físico y la maduración sexual y reproductiva, necesaria para
alcanzar el estadio de adulto. Por otro lado
muchas de sus tablas y figuras, son reconocidas por el nombre propio del
autor (como los estadios puberales que son utilizados universalmente). Cuando
Tanner publicó su trabajo se conocía poco sobre la regulación
neuroendocrina del eje hipotálamo-hipófisis-suprarrenal (HHG), tampoco utilizó la teoría de la evolución para la
interpretación de sus observaciones, pero aún así buena parte de su legado
clínico y de sus observaciones, permanecen[2].
Pubertad
y adolescencia, definición
Los términos pubertad y adolescencia son usados en
ocasiones de manera indistinta o intercambiables, pero no identifican a lo
mismo. Etimológicamente el término pubertad, proviene del latín “puber” (joven) y el sufijo tad (cualidad). Puber procede de la palabra latina pubis o púber,
que designa al vello púbico. Así que
etimológicamente pubertad significaría joven con vello púbico. La pubertad es el periodo que marca la metamorfosis que convierte el niño en
adulto.
Con más precisión, como veremos enseguida la pubertad es
el periodo de transición que va de la
“primera infancia” (chilhood) a la
adolescencia y coincide con lo que más
adelante, siguiendo a Hochberg Z. et
al,[3] llamaremos juvenility (“segunda infancia”)[4] y se identifica por la
aparición y desarrollo de los caracteres secundarios, un crecimiento acelerado
(estirón), cambios en el comportamiento y en ocasiones el inicio de la capacidad reproductiva. Es el
momento en el que se produce la
activación del sistema neuroendocrino hipotálamo-hipófisis- gonadal
que culmina con la maduración de las gónadas, la producción de las hormonas sexuales y el
comienzo de sus efectos biológicos.
Aunque hay diferentes signos físicos y bioquímicos con
los que se puede identificar el comienzo de la pubertad, todo el mundo sigue
los indicadores deTanner para niños y niñas,
basados en el momento en el que aparecen las mamas en niñas y cuando comienza a aumentar el volumen de
los testículos y del pene en niños, así como el vello público en ambos (Fig.
2).
En niñas el primer signo de comienzo de la pubertad es el
desarrollo de las mamas: Estadio 2 de Tanner (B2) o la aparición de vello
púbico (PH2), habiendo algunas diferencias individuales e inter-raciales: en
las chicas negras suele aparece primero el vello púbico mientras que en las
blancas suele iniciarse la pubertad con el crecimiento mamario[5].
Fig. 2. Estadios
puberales de Tanner

En los chicos el primer signo externo de pubertad suele
ser el incremento del tamaño de los testículos por encima de 3 c.c (ml) (Estadío Tanner 2 (G2)). El tamaño de
los testículos se suele medir utilizando la comparación con el orquidómetro de
Prader y es debido al crecimiento de los túbulos seminíferos estimulados por la
FSH.
Regulación
endocrina de la pubertad
La
pubertad tiene lugar como consecuencia de la activación de los
ejes hipotálamo-hipófisis-gonadal (HHG) e hipotálamo-hipófisis-adrenal
(HHA), un momento de la evolución corporal en el que se ponen
en marcha una serie de mecanismos estimuladores e inhibidores que funcionan
mediante una relación en feed back (Fig. 3).
Fig. 3.
Ejes Hipotálamo-Hipófisis- Gonadal (HHG) y eje Hipotámo-Hipófisis-Adrenal.
Tanto en niños como en niñas la pubertad tiene lugar cuando se activan los
mecanismos de feed-back de ambos ejes (ver texto)

SNC: Sistema Nervioso Central. Gn-RH(Gonadotropin
Relesing Hormone);
LH( Luteotropìn Hormone) y FSH
(Follicle-stimulating hormone):
CRH(
Corticotrophin Relesing Hormone; ACTH (Adreno Corticotropin Hormone); DHA(
Dehidroepiandrosterone)
En la
mayoría de los mamíferos el eje HHG está
activo en la segunda mitad del embarazo, en el periodo neonatal y en la
infancia temprana, provocando un aumento de los esteroides sexuales que participan en la diferenciación sexual y
en otros programas del sistema nervioso.
La actividad del eje HHG está
quiescente durante el periodo de la primera infancia (chilhood). Pasado el periodo prepuberal, es cuando se produce una reactivación importante
del eje, bajo la forma de un incremento de los pulsos hipotalámicos de GnRH (Gonadotropin-releasing
hormone) , lo que provoca un aumento de la gonadotropinas LH (Luteotropìn
Hormone) y FSH (Follicle-stimulating hormone). LH estimula la
maduración folicular en el ovario,
induciendo la síntesis de
estradiol en la capa glomerular y de andrógenos en las células tecales
ováricas, así como de progesterona en el cuerpo lúteo y de estradiol después de
la ovulación, así como de las inhibinas A y B,
Induciendo los ciclos ováricos que con la menarquía marcan el comienzo
de la actividad reproductiva en las chicas.
En los chicos la LH estimula la secreción de andrógenos (testosterona y
androstendiona), en las células de
Leyding del testículo, induciendo la espermatogésis, comenzando así la “gonadarquia”. La FSH
actúa en la capa granulosa del ovario y en las células de Sertoli del
testículo, promoviendo la gametogénesis. Una activación del eje HHG que ocurre
antes de que se visibilicen los signos externos de la pubertad como la
telarquia, la maduración ósea, el aumento del volumen de los ovarios y del
útero o la propia menarquía.
En todo caso son las hormonas sexuales las responsables
de la aparición de los signos físicos de la pubertad. De la telarquia
(aparición del desarrollo mamario), pubarquia (aparición del vello púbico),
gonadarquia (la producción de hormonas sexuales por las gonadas) . manarquia
(comienzo de la regla en las chicas) y
espermarquia (comienzo de la producción de esperma por los chicos), siguiendo
una secuencia que fue descrita canónicamente por Tanner (Ver atrás).
Lo sorprendente es que el lugar anatómico en el cerebro
donde está el centro de operaciones de
este eje, está representado por muy
pocas neuronas (unas dos o tres mil)
capaces de secretar el GnRH, dispersas en la eminencia media del hipotálamo. En un momento determinado,
comienzan a producirse algunas señales (ver más adelante) que estimulan la secreción
pulsátil de GnRH a un ritmo aproximado
de 90 por minuto que es drenada a la red del sistema vascular (sistema portal)
que conecta el hipotálamo con la
pituitaria anterior, estimulando la secreción de LH y FSH[6].
La pubertad se asocia también y de manera independiente
con la activación del llamado eje hipotálamo-hipófisis-adrenal (HHA), mediante
el cual se estimula la actividad de la enzima 17-α-hidroxilasa (17,20 lyasa),
resultando en un aumento de la producción de la dehidroandosterona (DHEA), dehidroandrosterona
sulfato (DHAS) y de la androstendiona. Este periodo llamado adrenarquia es la
causa del comienzo de la pubarquia , con
la aparición del vello del pubis, de acné
y de un peculiar olor.
La adrenarquia es un fenómeno exclusivo de los primates[7] y los mecanismos moleculares que inducen la
aparición de la pubertad no son bien conocidos aunque se presume del papel de
mecanismos genéticos y ambientales (ver más adelante).
El eje HHG funciona como un sistema feedback en el que hay una serie de ciclos o asas de
autorregulación ultracortas, cortas y largas. Hay un asa ultracorta de
autorregulación del promotor del gen de la propia GnRH-1[8], otra corta entre la
hipófisis (LH y FSH) y el hipotálamo (GnRH)
y un asa larga entre el hipotálamo, la hipófisis y las gónadas. Finalmente son los esteroides sexuales (EE y
AA) los que van a remodelar buena parte
de la fisiología, de la anatomía y del comportamiento que se producen durante la pubertad y la
adolescencia.
La
adolescencia, (etimológicamente adolescere =
crecer), identifica el periodo de maduración que proporciona
al niño los hábitos, el comportamiento y las habilidades sociales que le
incorporan a la vida adulta. Incluye la pubertad pero también el llamado estirón puberal y crecimiento, la
maduración cognitiva y cerebral, y los aspectos sociales relacionados con el
aprendizaje, la intimidad y el apoyo mutuo, así como la intensificación de
amistades preexistentes, el desarrollo de nuevas relaciones y habilidades biosociales.
Frente a la pubertad que es un proceso de maduración
compartido, aunque con distintos ritmos con otras muchas especies y con todos
los primates, la adolescencia es un
periodo genuinamente humano, pues no puede hablarse con propiedad de
adolescencia ni siquiera en los primates superiores.
El objetivo final de todo el conjunto que identifica la
adolescencia es conseguir de manera satisfactoria, la socialización y la
madurez reproductiva que identifica la vida adulta. Para ello es necesario que la maduración mental y
hormonal vayan unidas lo que debe alcanzarse mediante procesos altamente
complejos, interacciones y mecanismos de “feed back” entre el sistema nervioso
central y el sistema endocrino[9], así como la relación de
ambos con los agentes externos que se engloban bajo el epígrafe general de
medio ambiente (enviromment). La aparición de la pubertad está condicionada
biológicamente aunque pueden influir condicionamientos ambientales (ver más
adelante), mientras que la adolescencia está condicionada sobre todo social y culturalmente.
Frente a las teorías de Sigmund Freud y de Stanley Hall
sobre el carácter universal de las pautas de desarrollo y de conducta de los
adolescentes, en las primeras décadas del siglo pasado una serie de estudios de
campo, de sociedades primitivas de Nueva Guinea y Samoa, hicieron posible la
aparición de otros puntos de vista sobre la adolescencia. Fue el caso, por
ejemplo de los trabajos teóricos de Ruth Benedict, y las investigaciones de
campo en Samoa de Margaret Meat, que mostraron la importancia de la influencia
de las instituciones sociales y los
factores culturales en el proceso evolutivo del desarrollo humano
Así, por ejemplo, mientras que en las
sociedades más desarrolladas el paso de la infancia a la edad adulta suele
ser discontinuo. Llos niños no ven nunca un parto ni un acto sexual ni
tampoco cuando muere una persona y no es raro que las mujeres
tengan su primera menstruación sin saber de qué se trata. En cambio, en
sociedades primitivas el proceso evolutivo es continuo, los niños tienen acceso
a experiencias consideradas tabú en occidente y son considerados básicamente
iguales a los adultos; la sexualidad no es reprimida y se la considera natural
y placentera. Igual ocurre con el juego y el trabajo que en las
sociedades occidentales el paso del juego irresponsable al trabajo
responsable se produce en forma abrupta, lo que contribuye a incrementar los
conflictos en los adolescentes, mientras que en las sociedades menos
desarrolladas el trabajo y el juego no están tan separados. Por otro lado en la
mayoría de las sociedades primitivas el paso de la niñez a la edad adulta, es
un “rito de paso” lleno de contenido ritual y simbólico distinto para cada
cultura, mientras que en los países desarrollados este ritual ha desaparecido
casi completamente.
Los ejemplos sobre la distinta manera de iniciarse en la
vida adulta a lo largo de la pubertad y de la adolescencia podrían
multiplicarse. Si la adolescencia es el periodo en el que los humanos se preparan biológica (pubertad) y socialmente
para la vida adulta y reproductiva, las grandes diferencias culturales (también
individuales) pueden llegar a explicar
por sí mismas algunas de las respuestas, incluso (en parte) las biológicas, de este periodo.
Podríamos decir, en fin, que mientras que la pubertad ha
sido el motivo de atención y de estudio de la neuroendocrinología, la
adolescencia lo ha sido sobre todo de la sociología, la psicología, la
antropología e incluso la filosofía, como es el caso del filósofo Gustavo Bueno[10], quien llega a distinguir hasta 27 tipos o
modelos de adolescencia, desde las ceremonias
de los bosquimanos del Kalahari a las de ingreso en las academias militares
actuales, (imposibles aquí ahora de revisar). Una diversidad que complica la
elección de una definición univoca de adolescencia y su dificultad para ser
estudiada solo de una perspectiva científica, lo que no significa para Gustavo
Bueno que haya que asumir ante los diferentes tránsitos o tipos adolescentes un relativismo cultural
antropológico en el que se acepten acríticamente todas las iniciaciones
rituales, tal como las asumiría un “antropólogo de gabinete”(Bueno dixit) pues “no será lo mismo por ejemplo, la
iniciación adolescente a la “cultura jarrai”, de la bandas de barrios urbanos, o
de sectas religiosas (tipo “los niños de Dios”)
que la iniciación al terrorismo etarra, a la
delincuencia, o a la alienación (ante las que hay que tomar partido) o que las
iniciaciones de algunas culturas primitivas que no por extremas dejan de tener un distintivo carácter étnico.
Desde luego las adolescencias actuales de las sociedades abiertas y democráticas son
mucho más complejas (en cuanto a clase sociales, conocimientos profesionales, demografía,
etc) que los de las sociedades antiguas o ágrafas. Por ejemplo ahora en
nuestras sociedades se habla de “crisis
de la adolescencia” que es un concepto inconcebible en las segundas. Pero
hay que tener cuidado en no magnificar las diferencias, pues en la sociedad
actual son muy importantes los valores dominantes dentro de cada clase social.
Así, dice Bueno: “no pueden ser del mismo orden las «crisis de adolescencia» de
un príncipe o de un aristócrata, incluso del heredero de un agricultor rico
–todos estos sujetos tienen ya prefigurada, en cierto modo, su vida adulta, a
la manera como la tienen prefigurada todos los jóvenes de las tribus ágrafas
(precisamente será en estas clases en donde florecerán las ceremonias de
«puesta de largo», «de graduación», etc )– que las «crisis de adolescencia» de
los hijos de empleados urbanos, de trabajadores industriales que buscan la
«promoción social» de sus descendientes haciendo lo posible por darles estudios
y lograr su ingreso en la universidad”. Por otro lado, termina Bueno: “hablar
ahora de la «adolescencia» como «fase previa a la integración social» y de las
«crisis de la adolescencia» como «problemas psicológicos» comienza a ser una
forma de cultivar el humor negro”, en un momento en el que la ausencia de
perspectivas para muchos jóvenes prolonga la integración social y, por tanto,
de alguna forma la adolescencia, hasta
edades nunca antes vistas en la historia del hombre.
Esta situación entre una maduración sexual más precoz
(ver más adelante) y al mismo tiempo un retraso en el reconocimiento por parte
de la sociedad como individuo adulto e independiente, está marcando una situación nueva y paradójica en
nuestra especie, cuyas consecuencias en términos de morbilidad biológica,
psicológica y social estamos ahora comenzando
a valorar.
Aunque la maduración puberal y la de la adolescencia
suelen coincidir son procesos que
obedecen a mecanismos adaptativos en ocasiones diferentes, que cuando se
disocian demasiado en el tiempo pueden dar lugar a disfunciones, algunas con
repercusiones clínicas importantes.
¿Cuáles
son las señales que inducen al cerebro a producir pulsos de GnRH e iniciar la maduración
sexual?.
El objetivo final de esta transformación de niño a adulto
a través de la metamorfosis[11] puberal y de la
adolescencia es conseguir una satisfactoria capacidad reproductiva. La
reproducción y el resto de comportamiento que tienen que ver con la
supervivencia en la vida adulta son situaciones que exigen una gran demanda de
energía, que depende además del comportamiento de cada especie. Parece pues
importante que cuando tenga lugar el momento de la reproducción (y sus consecuencias
de defensa, alimentación, supervivencia, etc)
el cuerpo esté en las condiciones más satisfactorias para tener éxito.
El timing de la pubertad y de la
adolescencia es pues crítico.
Pero,
¿cuáles son las señales que le hacen al cerebro comenzar la cascada que
comienza con el aumento de la frecuencia de los pulsos de GnRH?
Hasta hace no demasiado, apenas nada era conocido sobre las señales que
inducen la maduración sexual y el comienzo de las potencialidades
reproductivas. Una cuestión difícil de investigar por su complejidad y
diversidad pues, entre otras cosas, puesto que las circunstancias son muy diversas las
señales varíen para cada especie.
Hoy sabemos que el penúltimo escalón que induce la pubertad es la supresión, en un momento
determinado de los estímulos nerviosos
centrales inhibidores sobre la GnRH,
dando lugar a una secreción episódica (pulsos) y tras ella el aumento de la
producción de LH y FSH, seguido del de los estrógenos en las chicas y de los andrógenos e
inicio o de la espermatogénesis en
chicos, iniciándose la cascadas que culminará con la madurez
reproductiva y social.
Son numerosas los
agentes que se han propuesto en los últimos tiempos, relacionados con el
comienzo de la pubertad.
El progreso sobre el “estado del arte”
en esta cuestión puede valorarse, por ejemplo, comparando dos revisiones sobre el tema una de
2004[12] y otra de 2017[13]. En los últimos años se han producido un
número importante de estudios que están comenzando a explicar cuáles son estos mecanismos
que regulan tanto las señales genéticas
como las que vienen del exterior y que
inducirían el comienzo de la pubertad. La Insulina[14], la Leptina, la Dopamina y la Prolactina que tienen un
efecto sinérgico sobre la secreción de GnRH[15], la Grelina [16] , el Glucagon-Like Peptid-1 GLP-1 y el Péptido YY3-36 (PYY)[17], o la Adiponectina[18] , con efecto inhibitorio. Inhibidores genéricos como el ácido
gamma-amino-burítico (GABA)[19] o estimuladores como el Glutamato[20], los péptidos opiáceos[21], la Melatonina[22], Óxido Nítrico (NO)[23], Ácido retinoico (Vitamina A)[24], IGF-1[25] o
determinados genes como el GPR54[26] (que codifica el receptor
de la Kisspeptina, o la Kisspeptin (el
péptido más potente liberador de la gonadotropinas, de entre los conocidos[27]) , codificado por el gen
KISS1, la Neurokinina B (NKB) un péptido que es co-secretado por algunas
neuronas que también secretan Kisspeptina, el gen CYP17 que controla la síntesis de
andrógenos y de los elevados niveles de estrógenos, o el Neuropétido Y (NPY)
que contribuye a la regulación de la secreción de GnRH, el gen relacionado con la proteína Agouti ,
ambos con efecto inhibitorio[28] el gen de la Proopiomelanocortina (POMC), de gran
importancia en la regulación del peso corporal, recientemente relacionado con la secreción de
la GnRH (estimulándola) [29], la Galanina-like peptide
(GALO)[30] (estimulando), el Steroided-
Factor 1 (SF1)[31],
la dopamina y la adrenalina (vía la tiroxina hidroxilasa)[32], son algunas de ellas. Recientemente el grupo de Tena Samper de Córdoba a partir de
estudios experimentales en roedores modificados genéticamente ha propuesto la existencia de un eje leptin--α-MSH--Kiss1—GnRH que estaría
envuelto en la regulación del efecto de la leptina en la maduración puberal, de gran importancia
para la regulación metabólica de la aparición de la pubertad [33] (Fig. 4)
A pesar del gran progreso de los
últimos años es esta una cuestión sobre la que se conoce aún poco, pues entre otras cosas
todas estas señales son “permisivas”, pero no son la causa de que se induzca la
aparición de la pubertad[34].(ver más adelante).
Fig. 4.
Mecanismos intermediarios relacionados con la secreción de Gn-RH

Por otro lado es bien conocido que los chicos y las chicas tienen un diferente “timing” en el desarrollo puberal y en la
adolescencia, aunque este ritmo varía entre especies, pues mientras que en humanos las chicas tienen una pubertad más
adelantada que los chicos, en otras especies ocurre lo contrario. Este
diferente ritmo puede estar controlado genéticamente pero es posible también
que tenga un origen fetal, dependiendo de cuándo y también de la intensidad de la
exposición de las hembras a los
esteroides sexuales[35].
También la adolescencia, tal como se ha definido arriba, -como
el conjunto de comportamientos y hábitos que llevan al chico o a la chica a la
vida adulta-, está sujeta a un reloj que, tal como ocurría con la
pubertad, puede estar controlado por
mecanismos relacionados con los esteroides o por otros independientes de ellos.
Numeroso estudios experimentales han demostrado como el comportamiento sexual
en los animales machos estaría condicionado por la testosterona y en las
hembras por el estradiol y la progesterona, que además no solo actuarían
estimulando la función sino remodelando las organización neural durante la
adolescencia (ver revisión en Sisk Ch L ..).
SITUACION NUTRICIONAL Y ESTRES
Desde luego el inicio de
la pubertad está marcado por un reloj
biológico, genéticamente condicionado,
pero también por las señales externas que
informan al hipotálamo de la situación del resto del cuerpo y también
del mundo exterior, de entre las cuales las nutricionales y metabólicas y las
asociadas al estrés, han mostrado ser críticas en el control de la pubertad.
Los mamíferos,
especialmente las hembras invierten una gran cantidad de energía para concebir,
transportar, cuidar y sacar adelante a su descendencia con el objetivo de
garantizar el mantenimiento de su linaje genético. Por esa razón tienen que asegurarse
de que cuando se produzca la maduración
sexual, el cuerpo esté lo suficientemente preparado para hacer frente a la
demanda energética. De igual manera en situaciones de dificultades en la
consecución de energía la función reproductiva tiende a suprimirse. Por esta
razón el eje HHG (y, ya veremos que también el eje HHA) debe recibir una
información integrada de la situación metabólica periférica. Así durante los periodos de estrés energético
los pulsos de GnRH llegan a suprimirse, reduciéndose la producción hipofisaria
de LH y FSH, induciendo a la postre una
hipogonadismo hipotalámico[36].
La
“plasticidad” corporal.
Se suele llamar
“plasticidad” a la capacidad de un determinado genotipo de producir
diferentes fenotipos como respuesta a diferencias en el medio ambiente[37]. La llamada tendencia
secular del crecimiento en los niños y de la pubertad (en los últimos 150 años)
es un buen ejemplo de esta plasticidad corporal.
Etapas
de la historia de la vida
Hace 3 o 4 millones de años, el homínido Australopitecus afaerensis tenía, como hoy le ocurre al chimpancé, solo
3 etapas entre el periodo inmediatamente postnatal y el del adulto: cinco años
de infancia (infancy), cinco años
juveniles (chilhood) y 2 años como joven (juvenile), antes de la aparición de la reproducción. Durante la
evolución con la aparición del homo
sapiens las etapas de la infancia y la adolescencia cambian. Desde la perspectiva del modelo evo-devo en el Homo sapiens es posible siguiendo a Hochberg
Z, et al, identificar cinco fases prolongadas y pronunciadas en su historia de
vida[38]:
Hochberg
Z, et al,)
|
Español
|
Definición
|
|
I
|
Infancy
|
Periodo postnatal y lactancia
|
30-36 semanas de vida
(1-2 años)
|
II
|
Chilhood[39]
|
Primera Infancia
|
Hasta los 2-6 años de vida
|
III
|
Juvenile (middle chilhodd)
|
Infancia media
(periodo prepuberal)
|
Un o una joven mientras es prepuberal. Puede durar otros 3-4 años
más, concluye con la maduración sexual en los niños y la menarquía en las
niñas (7 a -12-14 años)
|
IV
|
Adolescencia
|
Adolescence
|
Puede durar otros 3-5 años más
|
V
|
Youth
|
Jóvenes
|
Variable en función de las culturas y la situación social y
económica. Culmina con la paternidad a una edad media (mundial) de 20 años en
las mujeres y 24 en los hombres.
|
El paso de una etapa a otra del desarrollo, es la consecuencia de
cambios en los sistema de regulación endocrina. El paso de infancy (primera infancia) a
“chilhood”(segunda infancia), se
caracteriza por una aceleración de la actividad del eje GH-IGF1 (Growth Hormone- Insulin Growth Factor-1)
. La transición hacia la etapa juvenility
(infancia media) requiere el
aumento de la actividad de la capa reticular
de las suprarrenales y el aumento de producción de andrógenos
(adrenarquia). El comienzo de la pubertad y de la adolescencia implica una maduración del eje
hipotálamo-hipófisis-adrenal[41].
La
importancia del periodo prepuberal (juvenile stage)
Todos los mamíferos, incluidos los grandes simios pasan
directamente desde la lactancia (infancy)
(primera infancia) al periodo prepuberal
(media infancia) (juvenile stage) sin pasar a través de la chilhood (segunda infancia),
tal como se ha definido arriba. En los humanos no ocurre así. Por un lado la primera infancia aumenta unos
30 o 36 meses, la etapa prepuberal (media infancia) (juvenility) tiene una duración adicional de 2 a 4 años. Un periodo este,
(juvenility), que dura 3-4 años seguido por la adolescencia
que dura de 3-5 años y, finalmente, una etapa de joven que dura una media de 4
años [41]. Este periodo (juvenility) (infancia media o prepuberal) desde la perspectiva
evolutiva seria un periodo distinto de la historia humana de la vida con peculiaridades específicas en
términos endocrinológicos y de la composición corporal, de gran importancia a la hora de enfrentarse
a los cambios sociales y a la maduración psicológica. Un periodo que correspondería
a lo que los psicólogos llaman (middle
chilhood). Es la edad en la que el cerebro adquiere el tamaño definitivo
incluso aunque las neuronas no hayan alcanzado su completa madurez y cuando
aparecen los molares de la vida adulta. En las sociedades actuales es también
el momento en el que comienzan a ir al
colegio y a ocupar espacios hasta entonces reservados a los adultos, ya sean
sociales, alimentarios, etc. Es también el momento en el que desarrollan un
fuerte olor, que produce cierta aversión en el caso padre-hija o
hermano-hermana, pero no entre otras relaciones familiares, lo que se
interpreta como un mecanismo de protección frente al incesto[42].
Con todas las limitaciones de los restos fósiles, hay evidencias de que hace 1,900.000 años los
homínidos (Homo habilis) podrían haber añadido una nueva estrategia evolutiva y
añadido algunas fases a su patrón de crecimiento (estrategia en su “life-history” ), manteniendo un periodo postnatal
(infancy) como sus predecesores e incorporando dos periodos nuevos, primera
infancia (“chilhood”) que facilitaría un periodo de rápido
crecimiento y de estabilización del
crecimiento con gran dependencia del soporte familiar y tribal. Más
recientemente se habría añadido, hace unos 100.000 años, la etapa de la adolescencia con el típico estirón puberal y la irrupción de la pubertad sexual,
demorando así la etapa de juventud (“younth”)
como correspondería a una especie que tiene una vida larga (Fig. 5)
Para
algunos antropólogos y primatólogos, el estirón puberal es la mejor definición
operacional de la adolescencia, incluso aunque aparezca antes de los caracteres
sexuales secundarios en las niñas o
mucho después de la aparición de los cambios genitales en los niños[43]. De hecho no hay
evidencias de la existencia en los simios de un estirón del crecimiento durante
la adolescencia parecido al de los humanos[44]. Por otro lado también
durante la adolescencia se produce un acumulo de grasa en el cuerpo.,
especialmente en las niñas, pero mientras
que en las hembras de chimpancé la acumulación de grasa se hace de
manera amplia por todo el cuerpo, en las adolescentes humanas se produce una
acumulación llamativa de grasa en muslos, nalgas, y senos incluso aunque la adolescente sea
delgada. Unas características que no son solo señales sexuales de maduración
sexual y reproductiva, sino también reservas grasas para hacer frente a épocas
de escasez.
Fig.
5. Evolución de las etapas de la vida (de los prohomínidos al Homo
sapiens

La transición del periodo de la segunda infancia (chilhood)
a la etapa prepuberal (juvenility) se
caracteriza por el comienzo de la producción de los andrógenos adrenales (adrenarquia), la
aparición del “rebote adiposo”, la desaceleración del crecimiento [45], y la aparición de la
primera muela[46].
La adrenarquia debe
ser una adquisición reciente de la evolución pues solo los humanos y los
chimpancés tienen adrenarquia, con aumento en este periodo (juvenility) de los niveles sérico de DHEA y DHEAS[47]. Por el contrario otros
primares como los babuinos o los Rhesus no la tienen y apenas si se produce un
incremento en este periodo de los niveles de DHEA[48].
Una de las características de esta etapa (juvenility) es que, como se ve en la
gráfica ha permanecido relativamente
constante a lo largo de la evolución, especialmente comparada con otras etapas
de la historia de vida como es la edad de la maduración sexual[49]. Esto es especialmente relevante pues DHA en
humanos funciona como un neuroesteroide, que influye en la función neurológica
y en la maduración del estado anímico[50], [51]. DHAE tiene también otros
muchos efectos sobre el sistema inmune[52] o el desarrollo y el crecimiento somático [53].
Ese efecto de la DHAE sobre el sistema nervioso ha
llevado a algunos autores a postular que
este periodo en el que tiene lugar la adrenarquia, que es también en el que se define el tamaño
del cerebro y de todo el cuerpo en relación con los simios, es también el que
condiciona la mayor esperanza de vida de los humanos respecto a los monos y que
cambios en el “timing” de la adrenarquia podría también modificar la esperanza
de vida[54].
Al igual que otros organismos, también los humanos han
evolucionado para poder resistir y superar las dificultades del medio ambiente
para mantener el fitness (aptitud), aunque sea de manera subóptima. El
largo periodo madurativo de los humanos y la especificidad de las diferentes
etapas permite mediante ajuste largo de
los tiempos de tránsito entre una y otra,
una mejor adaptación a las situaciones cambiantes del medio ambiente.
Transición
del periodo postnatal (primera infancia) (infancy)
a chilhood (segunda infancia)
Después de la gradual desaceleración del crecimiento en
el periodo postnatal la velocidad de crecimiento se incrementa rápidamente
entre los 6 y 12 meses de edad como consecuencia del paso de la primera
infancia (infancy) a la segunda infancia (chilhood), tras lo que se produce una estabilización de la
velocidad de crecimiento. Un retraso en este tránsito entre infancy y chilhood,
tiene una importante influencia en la estatura final y es responsable al menos
del 50 % de los niños sanos que son remitidos al endocrino pediátrico por talla
baja[55], de manera que se calcula
que por cada mes de retraso del tránsito de la primera a la segunda infancia se
produciría un déficit de crecimiento de 0,4 cm en niños y de 0,5 cm en niñas a
la edad de 5 años[56].
Es este un periodo muy sensible a influencias ambientales
muy diversas, especialmente todas aquellas relacionadas con una alta demanda de
energía que restaría de la necesaria para un adecuado crecimiento. Es el caso
de los déficit nutricionales asociados a problemas socioeconómicos o secundarios a problemas intestinales[57].
Otros ejemplos de mecanismos adaptativos en estas etapas del desarrollo proceden de
los estudios de población de sociedades actuales cazadoras. recolectoras. En
estos estudios se ha utilizado el periodo entre embarazos como un marcador “subrogado”
de la transición de la infancy a chilhood[58] . Un mayor periodo entre
embarazos podría ser interpretado como
un mecanismo adaptativo a situaciones de carencia. En este contexto es presumible encontrar una
relación inversa entre la duración de
los periodos inter embarazos y el peso corporal medio de adulto, como de hecho
se ha observado (ver gráficas tomadas de Gawlik A, et al.) [59].
Fig.
6 y Fig. 7: Intervalo entre partos. La línea de regresión del peso corporal de adulto
como una función del tiempo entre partos (niños y niñas).


Transición
de la etapa “juvenility” (media infancia o prepuberal) a la adolescencia.
Como veremos más adelante la edad de la menarquía está
condicionada aunque débilmente por la genética (herencia)[60] , aunque parece, -a la luz de las observaciones sobre la
tendencia histórica de la maduración sexual- , que lo es más aun por
circunstancias ambientales como la altitud, la temperatura, la humedad, la
luminosidad y, sobre todo por el balance energético[61]. La transición hacia la pubertad y la
adolescencia, así como los ajustes sociales que conlleva, tienen un alto coste
energético[62].
Unas señales medioambientales y metabólicas
que serian reconocidas por sensores hipotalámicos y que llegado a un cierto punto (el nivel más
satisfactorio) inducirían la liberación
de pulsos de alta frecuencia de GnRH, que a su vez inducirían la transición de
la fase juvenily a la pubertad y
adolescencia[63].
Esta transición desde la etapa “juvenile” (prepuberal) a
la adolescencia es pues otro periodo de
transición de gran importancia para la adaptación “plástica”, durante la
maduración corporal.
Desde una perspectiva evolutiva dos estrategias podrían
ser consideradas como mecanismos adaptativos para conseguir la mayor eficiencia
reproductiva (fitness: n producto de las tasas de fertilidad y de supervivencia):
Por un lado, por ejemplo cuando falta la energía
procedente de los alimentos, podría
retrasarse la transición hacia la pubertad y adolescencia, esperando de esta forma una situación corporal más madura con la que,
probablemente, podría garantizarse una
fecundidad tardía más eficiente.
Por otro lado en situaciones precoces de estrés
psicosocial en donde es previsible que la vida futura de la mujer sea difícil y
la esperanza de vida menor, la transición se acortaría, adelantándose la pubertad y la adolescencia, probablemente
como un mecanismo que permitiría aumentar el tiempo y las posibilidades
reproductivas.
En el momento actual en el que hay confort alimentario y social, se esté
produciendo un acortamiento del periodo juvenile
(prepuberal o media infancia), con
una menarquía más precoz (y probablemente un alargamiento de la etapa
adolescente y de la etapa youth
(juventud). Eso puede ser la consecuencia de la mayor presencia de
contaminantes y disruptores endocrinos,
pero también puede ser interpretado como
una respuesta evolutiva que intenta rentabilizar todas las posibilidades de fecundidad y reproducción, aproximando a los extremos las
capacidades genéticamente condicionadas
de la historia evolutiva.
Evolución
de la edad de la maduración sexual
La población humana ha crecido exponencialmente, desde
hace unos 80.000 años en las que debieron de haber solo varios miles de humanos
y en el que la especie humana estuvo al borde la extinción hasta los 7,400
millones de habitantes que se superaron en diciembre de 2016. Este crecimiento
de la población ha incrementado de manera muy importante la oportunidad de mutaciones
genéticas y por tanto, en teoría, acelerado el ritmo de la evolución humana[64].
Si bien la edad de la maduración sexual y de aparición de
la menarquía tiene un fuerte componente hereditario[65],[66] la tendencia
secular de la maduración sexual, medida por la edad de la aparición de la
menarquía es una muestra de que el desarrollo puberal tiene una gran capacidad
plástica de adaptación a las condiciones ambientales.
Influencia de la
herencia en la edad de la menarquia y la maduración sexual
La edad de la menarquía está condicionada por factores
genéticos y ambientales, así como por la
interacción
entre ambos (ver más adelante). La
asociación entre la edad de la menarquia en gemelos, o la correlación
encontrada entre la edad de la menarquía entre las madres y las hijas, sugieren
que aproximadamente la mitad de las variaciones fenotípicas en el “timing” de
la menarquía en las chicas de los países desarrollados es debido a factores
genéticos[67],
[68], [69], [70], [71]. [72].
No obstante los
resultados sobre el papel de la herencia en la edad de la menarquía están lejos
de ser coincidentes[73] siendo en ocasiones difícil de separar la
influencia ambiental de la genética.
Un mayor implicación
genética se ha visto en los casos de
pubertad precoz, especialmente en aquellos con gran agregación familiar en los
que se ha identificado una estrecha asociación con mutaciones en algunos genes
como el kisspeptin y sus receptores, o el gen
MKRN3 u otros generes
relacionados con los mecanismos de regulación y secreción del GnRH como los genes TAC3, TACR3, LIN28B,
GABRA1 and NPY [74].
De especial interés son los estudios recientes que
encuentran que dentro de las mismas familias se producen con mayor frecuencia
casos de pubertad precoz y casos de pubertad retrasada lo que sugiere que ambas
situaciones son la expresión de un continuo en la respuesta genética o
ambiental relacionada con la disfunción en la generación de los “pulsos de GnRH[75], [76].
Factores
ambientales y su relación con la edad de la menarquia
Numerosos factores ambientales se asocian con la edad de
aparición de la menarquía. La latitud y la irradiación solar[77], la etnia [78], el estado de nutrición[79], [80], la actividad física[81], el estándar de vida o la ausencia de padre [82], el estrés psicosocial[83] y otros.
La
maduración sexual a lo largo de la historia:
Diferentes estimaciones sugieren que la edad de la
menarquía en el periodo pre-neolítico (hace entre 20.000 y 12.000 años pudo
estar entre los 7 y los 13 años y ya en
el neolítico (entre 12.000 y 50.000 años) entre los 7 y los 12 años. Es decir que en el
neolítico la maduración sexual se produciría incluso antes que en el momento
actual[84] (lo que coincide con la
edad de la menarquía en poblaciones actuales cuyas condiciones de vida son
parecidas a aquellas que aun viven en condiciones muy primitivas[85]).
Sin embargo
su carácter plástico y el carácter
dinámico de este periodo de la maduración hacen que las respuestas sean
complejas y no siempre fáciles de predecir. De hecho es bien conocido que
cuando los animales de experimentación son sometidos a unos programas de estrés
crónico de malnutrición, infectivos o de otro tipo, la respuesta suele ser la de retardar la maduración
sexual. Por otro lado, cuando los
animales viven en un estado optimo que
permite un adecuado crecimiento, la
maduración sexual no ocurre hasta que ha finalizado el desarrollo juvenil
completamente. Por el contrario cuando el estrés no es tan grande como para
arriesgar la supervivencia inmediata, el
desarrollo sexual se adelanta, aumentado
así la probabilidad de reproducción antes de que se produzca una gran
discapacidad o la muerte. Esta respuesta en U es especialmente clara en
relación con el estado de nutrición. En el caso de tener en periodos tempranos
de la vida tanto un bajo peso como un peso muy alto respecto a la
media, la tendencia es hacia una
maduración sexual más precoz, mientras que aquellas chicas con un bajo
incremento de peso en la infancia (chilhood) suelen tener la maduración sexual
retrasada. Es decir la respuesta en la
maduración sexual es bifásica dependiendo del contexto en el que el estrés se
produce[86], [87]. (Fig. 8)
Es decir cuando los animales inmaduros experimentan
tensiones ambientales severas como malnutrición o enfermedad, la maduración se
retrasa a menudo hasta que las condiciones mejoren y el crecimiento normal
pueda reanudarse. Por el contrario, cuando los animales son criados en
condiciones ideales que promueven un crecimiento rápido, los controles internos
aseguran que la maduración no ocurra hasta que el desarrollo juvenil esté
completo. Pero cuando el estrés contextual no es tan grande como para desafiar
la supervivencia, el desarrollo puberal se acelera, aumentando así la
probabilidad de reproducción antes de la muerte o la discapacidad.
Fig.
8. Modelo Evo-Devo. Respuesta bifásica en la maduración sexual, dependiendo del
contexto.

Esta sería también la razón por la que a lo largo de la Alta
y Baja Edad Media y a medida que aumentaba
la densidad de las poblaciones, y con ellas las
hambrunas y las epidemias, se
produjo un retraso de la edad de la
menarquía hasta los 16,5 años como aun ocurre en las adolescentes de grupos
poco privilegiados de los países industrializados[88].
En el caso de una escasa esperanza de vida, como ocurre
aun en algunas comunidades que viven en situación muy primarias, la otra
estrategia evolutiva consistiría en disminuir la edad de la maduración sexual,
teniendo en el caso de los Aeta de Filipinas, la edad de la primera
reproducción a los 10 años. Una reproducción temprana reduciría la probabilidad
de fallecer antes de tener la oportunidad de reproducirse. Es decir que una precoz fecundidad sería la
consecuencia de un mecanismo adaptativo a la situación de alto riesgo de muerte
precoz, en unas chicas que habrían reducido su talla como consecuencia del
corto periodo de crecimiento en la preadolescencia[89].
Evolución
de la edad de la menarquía
Las dificultades para evaluar los cambios en el tiempo de
la aparición de la pubertad y de la adolescencia han sido recientemente revisadas[90] .
La maduración sexual es un acontecimiento que varía entre
poblaciones y a lo largo del tiempo. En
la mayoría de los países europeos[91],[92],[93] así como en Israel[94], China[95] , Canadá[96] o USA[97] se ha visto que la edad
de la menarquía ha descendido a lo largo del siglo XX. Similares resultados han sido también
encontrados en países en vías de desarrollo[98].
Registros procedentes de diferentes países del norte de
Europa, particularmente, Noruega, Dinamarca y Finlandia muestran que la edad de
la menarquía ha decrecido desde los 16-17 años en el año 1797
hasta aproximadamente los 13 años
en el año 1981. Es decir en los
países industrializados, en los últimos 150 años la edad de la menarquía
ha descendido alrededor de cuatro años (Fig.9 y Fig. 10)[99].
Fig.9.
Descenso de la edad de la menarquía en los países occidentales entre 1840 y
2000

Fig.
10. Descenso secular de la edad de la menarquia

En España diferentes estudios también han confirmado esta
tendencia secular de la edad de la menarquia[100]. (Fig. 11)
Una tendencia general que en los últimos años parece
haberse detenido.
Fig.
11. Descenso de la edad de la menarquía en España

Un descenso también encontrado en la mayoría de los estudios
para la aparición de la telarquia, así como para la progresión de los estadios
B2 a B5, de Tanner[101].
Aunque el número de estudios realizados en chicos es
mucho menor, la mayoría de ellos llegan
a la conclusión de también existe un “secular trend” de la “gonadarquia”[102] .
La disminución de la edad de la menarquía y el incremento
de la estatura adulta se relaciona con la dieta y los cuidados de salud propios
y se asumen como signos de bienestar. Pero también este adelanto
de la menarquía, junto al de la telarquia y menarquía ha sido interpretado en
los últimos tiempos como resultado de la creciente presencia ambiental de
disruptores endocrinos que aceleran la
maduración del eje hipotálamo.hipofisarios [103].
Recientemente el papel de los contaminantes sobre la
pubertad ha sido extensamente revisado[104]. El panel de expertos sobre el que se
fundamenta la revisión en la que
incluyen numeroso estudios
experimentales, clínicos y epidemiológicos hechos en los últimos años concluye
que los estudios experimentales en animales y los estudios en humanos
realizados en humanos tanto sobre entidades clínicas (pubertad precoz,
pubertad retardada u ovario poliquístico, así como los estudios
epidemiológicos, apoyarían el posible papel de los llamados genéricamente
disruptores endocrinos de origen químico
junto al tamaño corporal, en la aparición o en la progresión de la pubertad,
tanto en chicos como en chicas. Sin
embargo la falta de concordancia de muchos estudios entre sí, la heterogeneidad
de los resultados sobre los diferentes marcadores de pubertad, los diferentes
resultados en chicos y en chicas, la dificultad de establecer los tiempos y las
dosis de exposición, la diversidad de agentes químicos, su ubicuidad, la
dificultad de diseñar estudios que contemplen el potencial disruptor de la mezcla de estos productos tal como se presentan en
el mundo real, etc, hace difícil el establecimiento de conclusiones definitivas
así como de estrategias de prevención.
Sin embargo sin negar el papel de los disruptores
endocrinos en el “timing” de maduración puberal, desde una perspectiva evo-devo, recientemente, otros autores[105], [106], [107] consideran que, por
ejemplo, el adelantamiento de la edad de
la menarquía y de la maduración sexual, no sería un estado clínico (una
enfermedad asociada a la contaminación actual) sino el resultado de mecanismos adaptativos a un
medioambiente actual más favorable.
En las sociedades antiguas la demora de la adolescencia y
del primer embarazo, permitiría que la joven hubiera adquirido un peso y corpulencia
suficiente para garantizar el éxito de la reproducción y una mayor
fertilidad.
Desde una perspectiva evolutiva la biología de la mujer
debería escoger entre garantizar el éxito reproductivo alargando la adolescencia
aun a riesgo de disminuir le vida fértil total y el de morir joven por
un medio ambiente hostil (hambruna, infecciones, etc.) acortando también de esta manera el periodo total
fértil.
“Evolucionary
theory of socialization”
En humanos la media de niños nacidos a lo largo de
numerosas poblaciones a lo largo de la historia es de 6-7 niños vivos. Aún hoy
muchas parejas pobres de países en vías de desarrollo sin medios
anticonceptivos solo consiguen ese número de niños vivos. Esta tasa de
fertilidad está muy por debajo de la capacidad humana de reproducción que puede
alcanzar entre 11 y 12 niños como se ha podido observar en algunas poblaciones
bien nutridas que no practican la anticoncepción como los Hutterites (en
español Huteritas[108].
En la Fig.12 vemos comparado con la población Hutterites (de
máxima fertilidad) la menor fertilidad de la población de mujeres inglesas y
escocesas de finales del siglo XIX. Es la consecuencia del acortamiento del
periodo de máxima capacidad reproductiva en la mujer, caracterizado por una
menarquía más tardía, un emparejamiento a más edad y una menopausia más precoz.
Fig.
12. Curvas comparadas de la capacidad reproductiva entre dos poblaciones (los
hutterites y la población de Inglaterra y Escocia).

Durante la segunda guerra mundial se produjo un
experimento social que solo más tarde
comenzaría a comprenderse en toda su magnitud[109]. Las niñas evacuadas de
sus casas de Helsinki como consecuencia de la guerra y enviadas a vivir a Suecia o Dinamarca, tuvieron la menarquía más jóvenes e incluso
tuvieron más niños que aquellas otras
chicas de la misma cohorte que permanecieron en sus casas y evitaron así el
trauma de separación de sus familias.
En los años noventa Belsky et al[110], propusieron un modelo o
teoría evolucionista de la socialización, en la que relacionaban las
situaciones de estrés psicosociales familiares (por ejemplo conflictos
maritales, abandono paterno, crianza difícil) o extrafamiliares (por ejemplo
bajos ingresos, desempleo) con una aceleración de la historia de vida (de las
cinco fases arriba descritas) y de las estrategias reproductivas (Fig. 13)
Fig.
13. Teoría evolucionista de la socialización (Belsky , 1991)

En condiciones de estrés psicosocial, familiar o extra
familiar, cuando la experiencia vital sugiere un mundo inseguro en el que las oportunidades de reproducción no están garantizadas, la selección natural favorecería
un desarrollo y maduración acelerada, pues en estas circunstancias un cuerpo inmaduro pondría en
un mayor riesgo a la mujer y a la posible descendencia, que una maduración precoz corporal. En este
sentido una maduración sexual y corporal
precoz podría tener más ventajas reproductivas que una maduración tardía. Una
teoría que coincide con las evidencias epidemiológicas de que un desarrollo
puberal precoz se asocia con un mayor riesgo sexual, primeros encuentros,
primeros besos, primeros acercamientos
genitales e intercambios sexuales y una
mayor tasa de embarazos en adolescentes[111].
La relación entre pubertad y maduración sexual precoz y
las situaciones psicosociales de estrés (autoritarismo parental, situación
familiar desestructurada) han sido confirmadas en posteriores estudios
longitudinales[112].[113].[114], situación que se asocia
con un mayor riesgo de prácticas sexuales de riesgo en la adolescencia.
Por los estudios recientes en modelos animales comenzamos
a saber que aquel “imprinting” de las
niñas trasladadas durante la segunda guerra mundial o los más recientes estudios sobre la
relación del estrés psicosocial con los “tiempos” de la maduración sexual, están probablemente bajo control epigenético[115]
Diferencias
individuales en la “plasticidad” del desarrollo
Como era de esperar hay una gran variabilidad en la
respuesta “plástica” del cuerpo a los estímulos socioculturales que condicionan
el “timing” de la maduración el
desarrollo sexual.
Esta gran variabilidad nos habla del factor biológico y
del condicionamiento genético en la maduración y el desarrollo sexual (ver
antes).
En estudios recientes de interacción
gen-medioambiente[116] se ha podido ver que la
mayor precocidad en la edad de la menarquía en las chicas con familias
conflictivas está condicionada por la variabilidad genética del gen del
receptor de estrógenos-α (ESR1). En estos estudios la calidad de la vida familiar correlaciono
positivamente con la edad de la menarquía
entre las participantes homocigotas para los alelos minoritarios de los dos polimorfismos estudiados (rs9304799, rs2234693) del gen. Pero no
entre las chicas con otros ESR1 genotipos
(Fig.14).
Fig.14.
Edad de la menarquia estimada a partir de la interacción entre el genotipo rs9304799 Fi y la calidad del ambiente
familiar (QFE).

Otros estudios no han encontrado relación alguna entre
los polimorfismos del gen del receptor y la edad de aparición de la menarquía[117] .
La
explicación epigenética
Numerosas observaciones han puesto de manifiesto como los
humanos (al igual que otras muchas especies) reducen su estatura y tamaño
corporal en situaciones de crisis energética, como una estrategia evolutiva de
adaptación a la crisis, aprovechando la capacidad “plástica” del periodo de
transición entre las diferentes etapas de crecimiento especialmente la de la
infancia a chilhood[118]. Hoy comenzamos a saber
que estas respuestas adaptativas son el
resultado de cambios en los mecanismos epigenéticos que afectan a la
programación de la velocidad de transición de una etapa a otra.
Cambios en las condiciones ambientales durante la
embriogénesis y en periodos muy precoces de la vida pueden alterar de manera
permanente la regulación epigenética que puede inducir un “imprintig” y una reprogramación del epigenoma,
influyendo en el crecimiento, en la composición corporal y en la transición de
unas etapas a otras de la maduración sexual y corporal. Unos mecanismos
epigenéticos que hoy por hoy son muy poco conocidos. Recientemente Bastiaan T.
Heijmans et al. [119] han estudiado la cohorte de personas que estuvieron
prenatalmente expuestas a la hambruna del invierno de 1944-45 (Dutch Hunger
Winter famine). Seis décadas después en aquellas personas que estuvieron
expuestas y en hermanos que no lo estuvieron
la metilación del DNA del gen de IGF2. Los resultados muestran por primera
vez en una gran cohorte una menor metilación en un gen relacionado con el imprinting (el gen del IGF2) en las personas
que estuvieron expuestas. La asociación fue específica para la exposición
periconcepcional (y no ocurrió por ejemplo en aquellos en los que la exposición
se produce en los últimos tiempos del embarazo), reforzando la idea de que el desarrollo
temprano de los mamíferos es un periodo crucial para establecer el imprinting epigenetico.
Fig.
15. Diferencias epigenéticas
persistentes asociadas a la exposición perinatal al hambre en humanos

Un trabajo posterior de los mismos autores ha mostrado
que los cambios en la metilación (hipo e hipermetilación) se producen en otros
genes[120]
. Los resultados sugieren también que
estos cambios dependen del sexo, de la exposición individual, además del
momento en la gestación de la
exposición.
Fig.
16. Diferencias en la metilación de genes candidatos entre expuestos y no
expuestos al hambre (Dutch Hunger Winter famine) (ver texto).

Un
ejemplo de plasticidad. Cambios en la composición y cantidad del tejido adiposo
en la pubertad. El caso particular del tejido adiposo marrón
La cantidad de grasa en el cuerpo humano es muy variada dentro de la vida de cada individuo y entre
individuos. Hace ya años que el tejido adiposo ha dejado de ser considerado
solo como un órgano de cubierta y protección y adquirido la categoría de uno de
los órganos endocrinológicos más activos y dinámicos del organismo. No es este
el lugar para hacer mayores precisiones a este respecto y existen numerosas y
excelentes revisiones al respecto[121], [122].
El crecimiento del tejido adiposo a lo largo
de la vida no es lineal.
El desarrollo del tejido adiposo blanco en la vida fetal
no ha sido muy estudiado. Estudios anatómicos sugieren que las primeras trazas de tejido adiposo son
detectables entre la semana 14 y 16 de la gestación desarrollándose después lentamente
de manera multilocular. Ya al principio del tercer trimestre el tejido adiposo
aparece con su carácter tisular pero en pequeña proporción[123].
Inmediatamente
después del parto el tejido adiposo representa aproximadamente el 14 % del peso
corporal e incrementa a los seis meses hasta representar un 25-30 % del peso corporal[124]. Este aumento es sobre
todo a expensas del tamaño de las células adiposas más que del número (ver más
adelante), posteriormente la mayor contribución al peso la representa la masa
libre de grasa (Fat Free Mass; FFM) (Si
bien durante chilhood la contribución
del FFM es similar en niños y niñas antes de la pubertad los niños acumulan más
FFM que las niñas, una situación que se mantiene y aumenta durante la pubertad).
La cantidad de grasa corporal es similar en niños y niñas
hasta los 3-5 años de edad y después a lo largo de toda chilhood
las niñas acumulan más grasa corporal que las niñas, una diferencia que aumenta
en la pubertad y la adolescencia[125] (Fig. 17).
Fig.
17.
FFM,
(Fat Free Mass) FM (Fat Mass) y porcentaje de grasa corporal
en niños y niñas europeos.americanos
desde la infancia hasta la vida adulta (edad de 20 años).

Si utilizamos como medida antropométrica el IMC (BMI
(Peso/T2), vemos como aumenta a lo largo del primer año de vida,
después decrece en la primera infancia (2-5 años y después aumenta en la
pubertad (Fig. 18).
Fig.
18. Evolución del IMC desde el nacimiento.

Estos cambios en la acumulación grasa varían entre
individuos habiendo sido descrito por Roland-Cachera que el precoz incremento de peso en la infancia tardía (rebote adiposo) tienen
una gran valor predictivo en la obesidad en la adolescencia[126].
Fig.
19. Rebote adiposo. Varios ejemplos tomados de Rolland- Cachera
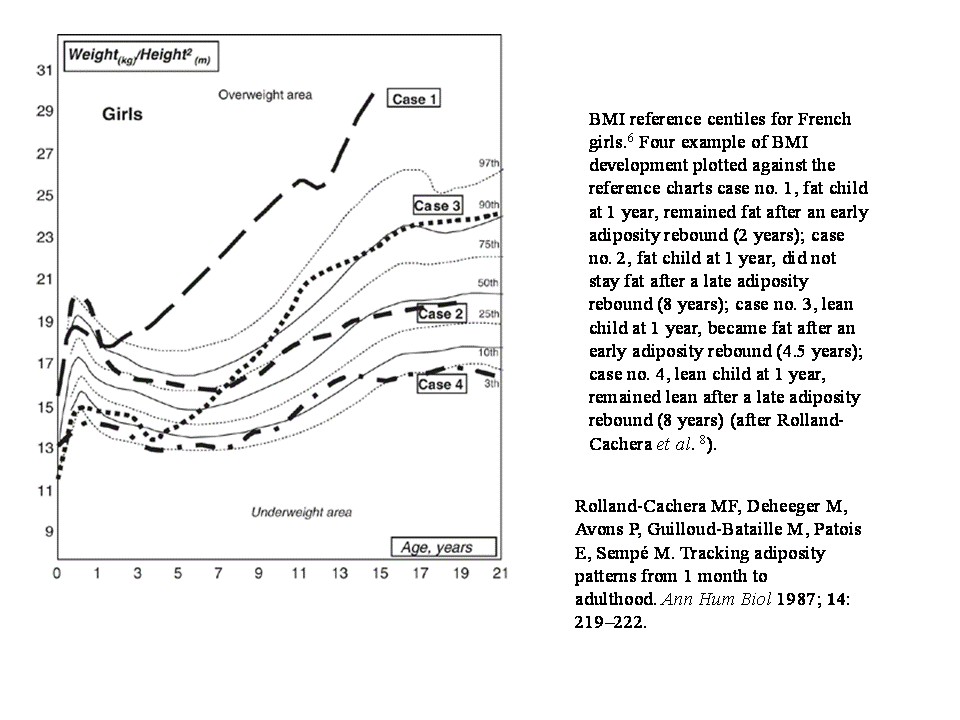
Este crecimiento del tejido adiposo en los dos primeros
años de vida es sobre todo a expensas del tamaño de los adipocitos más que del
número que comienza a aumentar a partir del segundo año de vida y continua de
manera ininterrumpida hasta la vida adulta[127] (Fig. 20).
Fig.
20. Crecimiento del tejido adiposo desde el nacimiento (tamaño y número de los
adipocitos)

Papel
de la dieta en la determinación del momento de la pubertad
Como se ha venido comentando, desde una perspectiva
evolutiva, la edad del comienzo de la maduración sexual y por tanto de la
posibilidad de reproducirse, ha estado sometida a fuerzas que han intentando
maximizar en cada momento la eficiencia reproductiva en función de las
circunstancias ambientales. El cuerpo
humano muestra una gran capacidad
plástica de adaptación en función de estas circunstancias y, de entre
ellas, la disponibilidad de energía y de alimento suficiente es una de las más
poderosas. Una adaptación que puede comenzar ya desde el propio embarazo y
continuar a lo largo de toda la infancia, pubertad y también a lo largo de toda
la vida reproductiva.
Charles Darwin[128] , [129] describió en los
animales domésticos la relación entre la alimentación y la capacidad
reproductiva. Ya entonces se había observado que los animales domésticos bien
alimentados y no sometidos a un trabajo intensivo tienen más descendencia que
los mismos animales salvajes, que el trabajo intensivo retarda el momento en el
que los animales domésticos alcanzan la capacidad reproductiva o que la
cantidad de alimentos afecta a la capacidad reproductiva de un mismo animal.
Recientemente Villamor E y Janse EC, han llevado a cabo
una extensa revisión sobre las relaciones entre la nutrición y el “timing” de
la maduración sexual y aquí nos
limitaremos a hacer solo un resumen de su extensa y completa revisión[130].
1. Estado de nutrición en el embarazo y
pubertad.
Los autores llegan a la conclusión de
que los estudios en los que se relaciona la situación nutricional en el embarazo,
(medido por marcadores, como la talla de las madres, el peso o el IMC) o el peso antes del embarazo, el crecimiento
intraútero (crecimiento intrauterino
retardado), la presencia de diabetes gestacional o los estudios en gemelos o en el orden de los
nacimientos, no son concluyentes.
.
2. Lactancia materna y pubertad .
Los autores revisan diferentes estudios realizados en diferentes partes del
mundo, algunos de ellos prospectivos. Algunos encuentran asociación positiva
otros negativa y otros ninguna asociación entre el momento de la maduración
sexual y el tiempo de lactancia materna.
3. Ingesta de proteínas en la dieta
infantil y pubertad.
Aunque no todos los estudios son coincidentes, la mayoría de los
revisados coinciden en que la ingesta de proteínas de origen animal, como la
leche y derivados, así como de proteínas procedentes de carnes rojas se asocia
con un adelanto de la pubertad y que, por el contrario, la ingesta de proteínas
de origen vegetal se asocia con un retraso de la maduración sexual. Los autores
no descartan que estas asociaciones puedan ser debidas a terceras variables no
contempladas como la concentración de fibra en la dieta o a la presencia de
otros nutrientes como las isoflavonas. En un estudio realizado por nosotros
hace años en la población escolar de Benalmádena, pudimos comprobar la
existencia de una asociación estadística entre la ingesta de frutos secos y la
edad de aparición de la menarquía, una asociación también encontrada en otros
estudios[131]
(Figuras.Tablas 21).
Fig.
Tablas 21. Asociación estadística entre la ingesta de frutos secos y la edad de
aparición de la menarquía


4. Grasas en la dieta y maduración sexual. Tras la revisión realizada por Villamor et al,
llegan a la conclusión de que los resultados de los estudios que han evaluado
la cantidad de grasa en la dieta con la maduración sexual son inconsistentes,
pues unos han encontrado una asociación positiva otros negativa y otros
ninguna. Similares resultados discordantes han sido encontrados cuando han sido
estudiada la relación entre la concentración de los tres más importantes grupos
de ácidos grados (saturados, monoinsaturados y polinsaturados) con la
maduración sexual.
5. También
con la ingesta de carbohidratos los resultados son inconsistentes.
6. Muchos
de las asociaciones encontradas entre los macronutrientes y la maduración
sexual podría ser debida al efecto de los microinutrientes. Diferentes estudios
han evaluado la asociación con la ingesta de calcio, magnesio, hierro, vitamina
A, vitamina D y algún otro. Si bien se
han encontrado asociaciones no siempre han concordado entre estudios y
necesitan de confirmación. Otro posible confoundig
puede proceder de la presencia de tóxicos presentes en los alimentos, con
efecto como disruptores endocrinos que en los estudios en animales se ha
demostrado que tienen un potente efecto sobre la maduración sexual. Son los
casos del Bifenil Policlorohidrato (PCB), el Bifenols A, o los los pftalatos
.
Efecto
del aumento del peso y del crecimiento precoz sobre la maduración sexual:
La nutrición en los niños afecta a los patrones de
crecimiento y hay evidencias que proponen que estos patrones pueden estar relacionados
con el momento de la maduración sexual. En un estudio realizado en 20183 niñas
en Brasil aquellas que mostraron un crecimiento más rápido entre los 19 y los
43 meses tuvieron más probabilidades de tener la menarquía antes de 12 años que
aquellas niñas con un crecimiento más lento[132] . Similares resultados
encuentran en un estudio con niñas
británicas en donde aquellas con una
mayor tasa de crecimiento entre los 2 y los 7 años tuvieron también la
menarquía antes[133]. En niños se ha realizado menos estudios pero
en uno realizado sobre 770 niños en
Filipinas aquellos con una mayor velocidad de crecimiento entre el nacimiento y
los 6 meses de edad tuvieron también una pubertad más adelantada con mayores
niveles de testosterona a los 15-16 años de edad[134].
Los resultados de estos estudios muestran la importancia
de garantizar un adecuado peso y velocidad de crecimiento durante la infancia,
también de la complejidad a la hora de establecer relaciones entre los
distintos alimentos y la maduración sexual, como se deduce de las relaciones
entre la dieta y la dinámica en la celularidad del tejido adiposo a lo largo de
la infancia.
Ácidos
grasos de la dieta y tamaño y número de
las células adiposas.
El periodo gestacional, los dos primeros años de la vida,
el rebote adiposo o la pubertad son momentos en los que se produce un cambio en
la dotación adipocitaria y en la acumulación grasa y, por tanto, potencialmente sensibles a influencias
externas o ambientales que pueden afectar a la dotación adipocitaria[135], [136],[137], [138], [139].
En estudios realizados por nosotros hace años pudimos
demostrar, no solo las observaciones anteriores sobre la dinámica de los
adipocitos en los primeros años de vida, sino también que en estos momentos existe una
estrecha relación entre el crecimiento adipocitario y los cambios en la
composición por el tejido adiposo de determinados ácidos grasos especialmente los ácidos grasos
n-6 que como es bien conocido no son sintetizados por el organismo (Fig. 22 y
Fig. 23)
Fig.
22 y Fig. 23. Concentración de ácidos grados en el tejido adiposo a lo largo
del crecimiento


Más recientemente en un
trabajo prospectivo realizado en 180 niños estudiados desde el momento del nacimiento
hasta los 24 meses de edad, pudimos comprobar que si bien, como era de esperar,
el IMC en el momento del nacimiento y a
los 12 meses estuvo correlacionado, la
probabilidad de que pasado el primer año el niño hubiese aumentado su IMC-
Z-score por encima de su IMC-Z-score basal es menor en los niños que habían
introducido el aceite de oliva en su
dieta habitual (frente a los que tomaron girasol) (36,4 % vs 60,6 %) (OR=0,37;
IC95%=0,15-0,87; p=0,02). La fuerza de esta asociación se mantiene después de
ajustar el modelo por la edad, el IMC al inicio del estudio o la actividad
física estimada por el número de horas que el niño permanece sentado[140]. Es decir, la dieta, la calidad de la dieta,
especialmente la composición grasa de la dieta,
influye ya de manera muy precoz en el distinto desarrollo graso (Fig.
24).
Fig.
24. Incremento de peso en los dos primeros años de vida en función del tipo de
aceite vegetal utilizado en la dieta,

No obstante los resultados del efecto de los ácidos
grasos sobre el crecimiento del tejido adiposo están aún lejos de ser concluyentes[141].
El peso corporal, como
resumen y conclusión de la situación metabólica es una de las señales mejor
conocidas desde hace tiempo. Y la obesidad uno de los problemas clínicos de los
que se conoce su asociación con la infertilidad y problemas en la maduración
sexual.
Peso corporal
y maduración sexual
Hoy está ampliamente
asumido que parte del adelanto de la menarquía que se está produciendo en todo
el mundo, especialmente en los países industrializados es debido a la mejora en
el estado de nutrición y al aumento de peso poblacional.
En el año 1987, llevamos a cabo un estudio en
niñas en edad escolar en la población de Benalmádena, que después comentaremos[142],
[143].
Con este motivo hicimos una revisión
sobre las relaciones entre el estado de nutrición y la dieta con la edad de la
menarquia[144]. En esta revisión recordábamos que fue en
1952, cuando Keneddy y posteriormente
Mitra y Widowson publicaron las
primeras observaciones en ratas acerca de que la malnutrición puede retrasar
tanto el crecimiento como el desarrollo sexual,
emitiendo la hipótesis de que la maduración sexual está estrechamente relacionada con el tamaño
corporal, de manera que las ingesta de alimentos o sus consecuencias
metabólicas puede actuar como una señal para inducir la pubertad[145],[146].
Estudios experimentales más recientes
en ratas a las que someten a diferentes modelos de desnutrición
encuentran que tanto en ratas macho como
hembras la malnutrición temprana influye en la aparición de la pubertad independientemente
del peso, llegando a la conclusión de que el
periodo perinatal es un momento
crítico para los cambios que condicionan
el timing de la pubertad[147].
En humanos ya Tanner había observado que para la misma edad son más altas y
pesan más las niñas que han tenido la menarquía, siendo el peso en el momento
de la menarquía independiente de la edad[148] Los estudios posteriores clínicos y
epidemiológicos la han ratificado., sobre todo los realizados por Rose Frich y
Revelle quienes establecieron el peso medio necesario para tener la menarquía
en 48 kilogramos [149].
Resultados similares fueron encontrados por nosotros en el estudio realizado en
la población escolar de Benalmádena en
791 niñas con edades comprendidas entre 8 y 16 años[150].
En este estudio realizado a mediados de los años 80 del pasado siglo la edad
media de la menarquía fue de 12,6 años y el peso corporal medio que se asoció
con la edad de la menarquía de 48 Kg,
exactamente el mismo que ya había sido encontrado por Rose Frich.(Fig.25 y Fig.
26 ).
Fig.25. Hipótesis de Frisch

Fig. 26. Edad de la menarquía en función del
peso corporal

Aunque el peso medio en el que aparece la
menarquia es bastante constante hay una gran variabilidad (entre 35 y 60 kg) lo que llevó a Frisch a
revisar su teoría proponiendo que más que un peso crítico existiría una
proporción critica de masa grasa respecto al peso (alrededor del 17%) y una mayor proporción
(alrededor del 22 % del peso corporal) para mantener la capacidad reproductiva [151].
Numerosos estudios
posteriores han confirmado esta relación entre el peso y la maduración
sexual. Es el caso de: “The National Heart Lung and Blodd
Institute´s National Growht and Healt Study, una cohort de 2379 niñas,
reclutadas en 1987 a la edad de 9 años y
seguidas con una frecuencia de un año. La edad media de la menarquía fue
de 12,7 años y el BMI para la misma edad fue consistentemente mayor en las
niñas que tuvieron la menarquia[152].
El Pediatric Research of Office Setting (PROS) Study, un estudio
transversal en el que el peso de las niñas fue estandarizado (Z Score) por el
peso medio de las tablas del NHANES III.
Como se ve en la figura, para todas las edades la niñas que tuvieron un
Tanner 2, su IMC estuvo siempre por encima del peso teórico esperado (Z>0)[153].
(Fig.27).
Fig. 27 Estadios de Tanner en función del
peso corporal

Una relación entre los
estadios de Tanner y la desviación respecto al peso teórico (Z Score) que
tendió a ser lineal como se ve en la Fig. 28:
Fig. 28. Relación entre el BMI y los estadios
de Tanner

En el Bogalusa Heart Study[154],
un gran estudio en el que se incluyeron 7 estudios transversales de niños entre
5 y 17 años de edad a lo largo de 20
años la edad temprana de aparición de la menarquía se
correlacionó negativamente con el BMI
tanto en niñas de raza blanca como negra
(r =-0,23 y r=-0,24) y la
incidencia de menarquia a una edad temprana (antes de los 11 años) fue mayor (1,75 veces mayor ) en niñas por
encima del percentil 75 del BMI que en niñas por debajo del percentil 25. (Fig.29).
Fig. 29. Proporción de niñas que han tenido la menarquia en diferentes edad, en
función de la raza, IMC y altura (Bogalusa Heart Study).

.
En niños la relación entre
el peso y la maduración sexual es más controvertida. En unos casos la relación
entre peso y maduración corporal se ha encontrado solo en niños[158]
o en ambos sexos [159], pero no en otros[160],
probablemente por la dificultad para
identificar los estadios de la pubertad en niños (la menarquía es un momento
fácil de identificar) o por el mayor protagonismo en la constitución corporal
del LBM en los niños. Otra posible explicación es que la relación entre el peso
y la maduración sexual no sea lineal y que pueda ser cierta para los niños con
sobrepeso, mientras que aquellos con obesidad severa la maduración sexual
podría ser más tardía[161].
¿Es el
incremento de peso, causa o efecto de la pubertad?
Numerosos estudios han
mostrado que los estrógenos y probablemente la progesterona favorecen la
acumulación de grasa (lo contrario ocurre con los andrógenos)[162]
. Varios estudios longitudinales, sin embargo parecen apoyar la tesis de que es el incremento de peso el que favorece la precocidad en la
aparición de la menarquía. He Q, Karlberg J et al., en un estudio con una amplia cohorte en Suecia, encuentran una correlación negativa entre los
cambios en el BMI entre los 2 y los 8 años de edad y el pico de máxima
velocidad, como marcador puberal. Los autores llegan a la conclusión de que el sobrepeso entre los 2 y los 8 años
puede no ser beneficioso para la talla final
pues el incremento temporal de talla en la infancia (chilhood) puede ser compensado con una
mayor precocidad puberal y una menor ganancia
de talla en la adolescencia [163].
En otro estudio diseñado
con el objetivo de investigar la dirección causal de la relación entre el peso
y el momento de la pubertad, Kirsten Krahnstoever Davison et al[164], estudian
una muestra de 183 niñas entre los 5 y los 9 años. Los autores miden desarrollo mamario,
niveles de estrógenos y el desarrollo puberal
llegando a la conclusión de que las niñas con mayor peso en la infancia
(early
chilhood) tuvieron más probabilidades de mostrar signos precoces de
pubertad, indicando que el estatus ponderal, precede al estatus puberal).
Resultados similares han
asido encontrados en otros estudios longitudinales recientes[165].
La leptina y la insulina
son dos de las hormonas que se han postulado como mediadoras entre el tejido
adiposo y la regulación del eje HHG. La leptina es una hormona secretada
por el tejido adiposo y se relaciona
estrechamente con su masa , teniendo
efectos muy importantes en la regulación del eje HHG[166],(ver
antes) por lo que no es sorprendente que
numerosos estudios tanto en roedores, como en primates o en humanos, hayan investigado el papel mediador de la
leptina en la regulación del timing
de la pubertad .
Por otro lado es bien
conocida la asociación entre obesidad e Iinsulinresistencia (IR), siendo
numerosos los estudios que han investigado el papel de la insulinoresistencia
en la patogénesis de la pubertad o en el desarrollo del síndrome de Ovario
Poliquístico (S.O.P) en la adolescencia[167]
.
En todo caso la
hipótesis de Frisch debe ser considerada en el contexto en el que se produce la
pérdida o la acumulación de grasa. Así, las niñas que se someten a un ejercicio
físico muy intenso (p.e deportes de élite o bailarinas) y que tiene muy poca grasa, tiene la
menarquía más tarde[168].
Mucha de la información arriba resumida ha sido obtenida
de la reciente y excelente revisión llevada a cabo por Kaplowitz PC et al.[169]
Los autores llegan a las siguientes conclusiones:
1.
Con los
estudios epidemiológicos llevados a cabo
en los últimos 30 años, se puede afirmar que
hay un sólida asociación entre el peso (medido por el BMI) y la
precocidad en la pubertad, medida sobre todo en las chicas por la menarquía,
aunque también probablemente con otros marcadores con la pubarquia o la
telarquia.
2.
Hay datos
sólidos que soportan la tesis de que el
incremento del peso corporal es el que antecede a la precocidad puberal y
no a la inversa,
3.
La leptina
puede ser uno de los eslabones
intermediarios entre el aumento de la masa grasa y el inicio de la
pubertad, pero que solo el incremento de leptina no es suficiente.
4.
Aunque el
aumento de peso no sea la única
causa del adelanto de la pubertad
observado a lo largo de los últimos años, sí que hay una relación entre el “secular trend” del peso y el secular trend de la pubertad.
5.
Esta
relación parece ocurrir también en niños pero que dadas las dificultades en
medir con mayor precisión el comienzo de la pubertad en niños así como el
distinto comportamiento de los compartimentos corporales en niños y en niñas a
lo largo de la pubertad, en niños estas relaciones son menos claras.
6.
Parece
necesarios estudios más contemporáneos pues los actuales comenzaron hace
décadas en un momento en los que la
prevalencia de obesidad era menor que ahora, donde se utilicen marcadores de pubertad y de
obesidad, tanto en niños como en niñas, menos subjetivos, que se amplíen el
rango de edad de los niños al menos entre 6 y 14-15 años y sobre todo estudios longitudinales que
incluyan, incluso, el periodo del embarazo, así como información sobre posibles
contaminantes ambientales y sobre todo sobre de ingesta alimentaria tanto
cuantitativamente (energía) como cualitativamente (ingesta de algunos alimentos en particular).
Estrés y maduración sexual
También la activación
del eje Hipotalámico-Hipófisis-Adrenal
(HHA) puede ser otra vía que conduzca a problemas relacionados con la actividad
reproductiva y con la maduración sexual. Es el caso del estrés inmunitario, asociado, por ejemplo
a las infecciones o la inflamación, el estrés psicológico, físico o
ambiental. En todas estas circunstancias
se puede producir una activación del eje HHA y asociarse con problemas en el
sistema de reproducción[170]. Sin embargo es posible que el efecto del
estrés sea percibido como señales de menor utilización de energía no siendo
posible separar ambas señales, habiéndose
propuesto una vía común que desencadenaría los trastornos del sistema
reproductor. La activación de la respuesta al estrés estimularía la producción
de glucocorticoides y de catecolaminas, que son hormonas contrareguladoras
del consumo de energía, generando señales de un estado
“hipometabólico”. El estrés tiene un
alto coste metabólico e induce mecanismos de respuestas similares a la de una
situación de baja utilización de energía. Ambos ejes no son independientes ni
tampoco las situaciones de estrés son puras por lo que es posible que lo más
frecuente es que hayan un efecto sinérgico entre el estrés nutricional y no
nutricional[171].
En
resumen: El estado de adulto (social y
reproductivamente hablando) necesita el paso de una maduración gonadal
(puberal) y social (adolescencia) a los que se llega a través de un
satisfactorio desarrollo neuro-hormonal cuyo “timing” está modulado por una serie de pistas y señales externas e
internas. El cerebro, en respuesta a estos estímulos internos y externos,
inicia la activación del eje HHG-GnRH, provocando un aumento de los esteroides
sexuales, que a su vez modulan la secreción de GnRH en el cerebro, al mismo
tiempo que se reorganizan circuitos neuronales que modulan el comportamiento
ante la reproducción y ante la vida
social del adulto.
Normalmente tanto la maduración gonadal, como la del
comportamiento (pubertad y adolescencia) aunque tienen mecanismos autónomos de
regulación están generalmente coordinadas desde el punto de vista temporal,
maximizando así el éxito reproductor y de supervivencia en este periodo
crítico. Este periodo es crítico, entre
otras cosas para la reorganización de la estructura y función cerebral lo que podría explicar que las diferencias
individuales en el comportamiento de la vida adulta podrían ser debidas en parte a las variaciones normales en la coordinación
entre la maduración gonadal (pubertad) y
la del comportamiento (adolescencia). El desarrollo del cerebro es particularmente susceptible a cambios en este periodo
acelerado en donde se puede hablar de un “estirón” también cerebral y no solo
de la talla. En humanos una disociación entre la maduración gonadal (pubertad)
y la adolescencia (social y de comportamiento) es muy probable que tenga
consecuencias importantes en el comportamiento el adulto. Es el caso, por ejemplo de los “eating
disorders” o del ejercicio extremo, que
significativamente retrasan la maduración sexual y con ella la exposición del cerebro a los
esteroides sexuales, o en el otro extremo el caso de la pubertad precoz que
índice a una exposición precoz del cerebro a los esteroides sexuales.
Como se ha visto en esta revisión la regulación del
sistema neuroendocrino que controla la maduración sexual y la fertilidad es muy
compleja, pudiendo muchas señales nutricionales o de estrés influir sobre esta
regulación. El sistema se caracteriza por la multiplicidad de señales y de
hormonas que intervienen, muchas de ellas redundantes. De entre ellas
solo la leptina parece ser imprescindible, no así la insulina o la grelina que
actúan más bien de manera” permisiva” para que intervengan otros actores.
En la Fig. 30 tomada de
Evans et al. Se resume las vías más importante a través de las cuales se
expresan las señales nutricionales bien estimulando (círculos grises), bien
suprimiendo (círculos negros) el eje HHG. Las líneas continuas representan vías requeridas de manera
imprescindible para la reproducción, las
vías con trazado discontinuo representan
vías críticas pero no imprescindibles. Las vías aquí no representadas
por líneas (p.e la grelina, AGPR/NYP,
Agouty/neuropeptido Y, Dopamina, Galanina, Kiss/Kissepetina, , NOS, POMC/CART
(proopiomelanocortina/cocaína , SF1( Steroidogenic factors-1VTA (Ventral
tegmental area), no son críticas para la regulación del eje.
Fig.
30. Resumen de las vías más importantes a través de las cuales se expresan as
señales nutricionales que regulan el eje HHG
(Hipotálamo-Hipófisis- Gonadal)

Tejido
adiposo marrón y pubertad
Más recientemente se le ha prestado una especial atención
al tejido adiposo marrón y no querríamos terminar esta revisión sin hacer
algunos comentarios sobre él pues es un
tema de gran actualidad del que aún se conoce poco, especialmente sobre su
dinámica a lo largo del crecimiento y el papel que pueda desempeñar en los
mecanismos adaptativos, especialmente en el periodo que nos ocupa de la pubertad y la adolescencia,
pues, entre otras cosas, las técnicas
para su estudio in vivo son muy recientes y aun no suficientemente
desarrolladlas. Rogers NH et al. han revisado
el “estado del arte” del tejido adiposo marrón en la pubertad[172]. Las células del tejido
adiposo marrón son muy abundantes en mitocondrias, acumulan múltiples gotas de
grasa y están definidas por la producción de UCP1 (Uncoupling protein-1) que es
capaz de estimular la producción de ATP a partir de la fosforilación oxidativa,
facultando la producción de calor y la
termogénesis.
Hasta hace muy poco se creía que en humanos el tejido
adiposo marrón existía solo en el último periodo del embarazo y en la primera
infancia, desapareciendo después. Sin
embargo estudios más recientes han demostrado la presencia de tejido adiposo
marrón a lo largo de toda la vida del
individuo. Los lugares donde el tejido
adiposo marrón se acumulan en mayor o menor intensidad son en la región
cervical, supraclavicular, interescapular, así como en axilas, mediastino y
zonas paravertebrales, perirenales y paraórticas.
En el nacimiento el tejido adiposo marrón puede suponer
hasta el 5 % del total del peso corporal, alcanza su mayor presencia alrededor de las 8 semanas de
vida y luego declina a lo largo de la primera infancia (chilhood) para aumentar de nuevo en el momento de la pubertad.
Diferentes estudios parecen sugerir que la cantidad de tejido adiposo marrón en
la etapa pediátrica y en la pubertad se asocia estrechamente con la edad y con
la maduración sexual (estadios de Tanner) , pero es independiente del BMI, del sexo o de la
estación del año (Fig. 31).
Fig.
31. Tejido adiposo marrón durante la adolescencia

De mayor interés es la observación de que los niños que
tienen presencia identificable de tejido adiposo marrón, tienen también una mayor masa muscular (y
ósea)[173],
una correlación que se observa en la pubertad donde se produce también un
aumento de la masa muscular. Un crecimiento sincrónico entre ambos tejidos
paralelo a los cambios en GH (Growth
Hormone) , esteroides gonadales, insulina, IGF-1(Insulin Growth Factor -1). También los glucocorticoides, las hormonas tiroideas, la leptina y las
catecolaminas intervienen activamente en la regulación del TAM. Habiéndose
propuesto a una molécula la irisina
como el nexo entre el tejido muscular y el tejido adiposo marrón.
Fig.
32. Evolución del tejido adiposo marrón a lo largo de la vida

La transición desde la adolescencia a la vida adulta se
acompaña de una disminución de la cantidad de tejido adiposo marrón, incluido
el tejido adiposo marrón estimulado por el frio, considerándose que solo lo
presentan un 5 % de las personas adultas frente a un 15-50 % de los menores de
18 años (aunque no se han hecho suficientes estudios en niños estimulados con
frio se presume que alrededor del 100 % % de los niños tendrían tejido adiposo
marrón tras el estimulo). En la mayoría de las personas ancianas el tejido
adiposo marrón no se detecta lo que tal vez podría explicar la mayor
intolerancia al frio de estas personas. También en las personas obesas, tanto púberes
como adultas, la detección del tejido adiposo marrón
estimulado por el frio es menor, lo que vincula al TAM con la fisiopatología de la obesidad (Fig. 32).
Finalmente el TAM a través de su efecto termogénico, se
ha asociado con conocidos factores de riesgo, como la acumulación de grasa
intratisular, la obesidad[174], la menor secreción de
insulina, la glucoregulación o el metabolismo lipoproteico[175], [176]. (Ver Fig. 33 y Fig. 34)
tomadas de los trabajos de Cypess y Chalfant).
Fig.
33. Tejido adiposo marrón en adultos humanos

Fig.
33. Asociación inversa
entre la activación del tejido adiposo marrón y la acumulación grasa.

[1] Tanner JM. 1962.
Growth at adolescence. Oxford:Oxford University Press.).
[2]
Ellison PT, Reiches MW, Shattuck-Faegre H, Breakey A, Konecna M, Urlacher S,
Wobber V. Puberty as a life history transition. Ann Hum Biol. 2012
Sep;39(5):352-60
[3] Hochberg Z, Belsky J.
Evo-devo of human adolescence: beyond disease models of early puberty. BMC Medicine. 2013;11:113.
doi:10.1186/1741-7015-11-113
[4] En este
capítulo (ver más adelante) utilizamos para identificar las etapas desde el
nacimiento a la juventud y vida adulta infancia a la adolescencia, los
criterios de Hochberg, et al.
Hochberg Z, Belsky J. Evo-devo of human adolescence:
beyond disease models of early puberty. BMC Medicine. 2013;11:113.
[5] Euling SY, Herman-Giddens
ME, Lee PA, Selevan SG, Juul A, Sørensen TI, Dunkel L, Himes JH, Teilmann G, Swan SH. Examination
of US puberty-timing data from 1940 to 1994 for secular trends: panel findings.
Pediatrics. 2008 Feb;121 Suppl 3:S172-91
[6] Plant TM, Barker-GibbML. 2004.Neurobiological
mechanisms of puberty in higher primates. Hum Reprod Update. 10:67–77.).
[7] Conley AJ, Pattison JC, Bird
IM. Variations in adrenal androgen production among (nonhuman) primates. Semin
Reprod Med. 2004;22(4):311–326
[8] Han YG, Kang SS, Seong JY, Geum D, Suh YH, Kim K. Negative regulation of
gonadotropin-releasing hormone and gonadotropin-releasing hormone receptor gene
expression by a gonadotrophin-releasinghormone agonist in the rat hypothalamus.
J Neuroendocrinol 11, 195–201.
[11] Empleamos aquí la palaba
metamorfosis como metáfora útil, pero no como referencia al sentido con el que
Freud la usa en “Metaformosis de la Pubertad. Tres ensayos de teoríasexual
(1905) en sus ensayos
[12] Sisk CL, Foster DL: The neural
basis of puberty and adolescence. Nat Neurosci 2004, 7:1040-1047
[13] Evans MC, Anderson GM.
Neuroendocrine integration of nutritional signals on reproduction. J Mol
Endocrinol. 2017 Feb;58(2):R107-R128.
[14] Kim HH, DiVall SA, Deneau RM
& Wolfe A (2005) Insulin regulation of GnRH gene expression through MAP
kinase signaling pathways. Mol Cell Endocrinol 242, 42–49.
[15]
Henderson HL, Townsend J, Tortonese DJ. Direct effects
of prolactin and dopamine on the gonadotroph response to GnRH. J Endocrinol.
2008 May;197(2):343-50.
[16]
Kluge M, Schussler P, Schmidt D, Uhr M & Steiger A 2012 Ghrelin suppresses
secretion of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) in
women. Journal of Clinical Endocrinology and Metabolism 97 E448–E451
[17] Pinilla L,
Fernandez-Fernandez R, Vigo E, Navarro VM, Roa J, Castellano JM, Pineda R,
Tena-Sempere M & Aguilar E 2006 Stimulatory effect of PYY-(3-36) on
gonadotropin secretion is potentiated in fasted rats. American Journal of
Physiology: Endocrinology and Metabolism 290 E1162–E1171
[18]
Klenke U, Taylor-Burds C & Wray S 2014 Metabolic influences on
reproduction: adiponectin attenuates GnRH neuronal activity in female mice.
Endocrinology 155 1851–1863
[19]
Zhu J, Xu XH, Knight GE, He C, Burnstock G, Xiang Z. A subpopulation of
gonadotropin-releasing hormone neurons in the adult mouse forebrain is γ-Aminobutyric
acidergic. J Neurosci Res. 2015 Oct;93(10):1611-21.
[20]
An XF, Niu YF, Ten SC, Liu JS, Feng H, He M, Shen XJ. Orphanin FQ and glutamate
connection in the regulation of gonadotropin-releasing hormone secretion in the
preoptic area of conscious male rats. Neurosci Lett. 2008 Aug 1;440(2):109-12.
[21] Grachev P, Li XF, Kinsey-Jones JS, di
Domenico AL, Millar RP, Lightman SL, O'Byrne KT. Suppression of the GnRH pulse
generator by neurokinin B involves a κ-opioid receptor-dependent mechanism.
Endocrinology. 2012 Oct;153(10):4894-904
[22]
Alvarado MV, Carrillo M, Felip A. Melatonin-induced changes in kiss/gnrh
gene expression patterns in the brain of
male sea bass during spermatogenesis. Comp Biochem Physiol A Mol Integr
Physiol. 2015 Jul;185:69-79.
[23]
Bonavera JJ, Sahu A, Kalra PS & Kalra SP (1993) Evidence that nitric oxide
may mediate the ovarian ste- roid-induced luteinizing hormone surge:
involvement of excitatory amino acids. Endocrinology 133, 2481–2487.
[24]
Cho S, Chung JJ, Choe Y, Choi HS, Han KD, Rhee K & Kim K (2001) A
functional retinoic acid response
element (RARE) is present within the distal promoter
of the rat gonadotropin-releasing hormone (GnRH)
gene. Brain Res Mol Brain Res 87 , 204–21.
[25]
Hiney JK, Srivastava V, Nyberg CL, Ojeda SR & Dees WL (1996) Insulin-like
growth factor I of peripheral origin acts centrally to accelerate the
initiation of female puberty. Endocrinology 137, 3717–3728.
[26]
Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno JS Jr, Shagoury
JK, Bo-Abbas Y, Kuohung W, Schwinof KM, Hendrick AG, Zahn D, Dixon J, Kaiser
UB, Slaugenhaupt SA, Gusella JF,
O'Rahilly S, Carlton MB, Crowley WF Jr, Aparicio SA, Colledge WH. The GPR54
gene as a regulator of puberty. N Engl J Med. 2003 Oct 23;349(17):1614-27
[27]
George JT, Seminara SB. Kisspeptin and the hypothalamic control of
reproduction: lessons from the human. Endocrinology. 2012 Nov;153(11):5130-6.;
[28] Roa J & Tena-Sempere M
2014 Connecting metabolism and reproduction: roles of central energy sensors
and key molecular mediators. Molecular and Cellular Endocrinology 397 4–14.
[29] Xu, Y., Faulkner, L.D., Hill, J.W., 2012. Cross-talk
between metabolism and reproduction: the role of POMC and SF1 neurons.
Frontiers in Endocrinology 2: 98.
[30] Shioda S, Kageyama H, Takenoya F & Shiba K 2011
Galanin-like peptide: a key player in the homeostatic regulation of feeding and
energy metabolism? International Journal of Obesity 35 619–628.
[31]
Tong QC, Ye CP, McCrimmon RJ, Dhillon H, Choi B, Kramer MD, Yu J, Yang ZF,
Christiansen LM, Lee CE, et al. 2007b Synaptic glutamate release by
ventromedial hypothalamic neurons is part of the neurocircuitry that prevents
hypoglycemia. Cell Metabolism 5 383–393.
[32]
Konner AC, Hess S, Tovar S, Mesaros A, Sanchez-Lasheras C, Evers N, Verhagen
LA, Bronneke HS, Kleinridders A, Hampel B, et al. 2011 Role for insulin
signaling in catecholaminergic neurons in control of energy homeostasis. Cell Metabolism 13 720–728
[33] Manfredi-Lozano M, Roa J,
Ruiz-Pino F, Piet R, Garcia-Galiano D, Pineda R,
Zamora
A, Leon S, Sanchez-Garrido MA, Romero-Ruiz A, Dieguez C, Vazquez MJ,
Herbison AE, Pinilla L, Tena-Sempere M. Defining a
novel leptin-melanocortin-kisspeptin pathway involved in the metabolic control
of puberty. Mol Metab. 2016 Aug 11;5(10):844-57.
[34]
Sisk CL (Op.cit)
[35] Sisk CL (Op.cit)
[36] Castellano JM, Navarro VM, Fernandez-Fernandez
R, Nogueiras R, Tovar S, Roa J, Vazquez MJ, Vigo E, Casanueva FF, Aguilar E, et
al. 2005 Changes in hypothalamic KiSS-1 system and restoration of pubertal
activation of the reproductive axis by kisspeptin in undernutrition. Endocrinology
146 3917–3925
[37] Hochberg Z, Feil R, Constancia M, Fraga M,
Junien C, Carel JC, Boileau P, Le Bouc Y, Deal CL, Lillycrop K, Scharfmann R,
Sheppard A, Skinner M, Szyf M, Waterland RA, Waxman DJ, Whitelaw E, Ong K,
Albertsson-Wikland K. Child health, developmental plasticity, and epigenetic
programming. Endocr Rev. 2011 Apr;32(2):159-224.
[38] Gawlik A, Hochberg Z. Lessons from the life history of natural
fertility societies on child growth and maturation. Swiss Med Wkly. 2012 Jun 19;142
[39] El término childhood es poco específico e implica un amplio rango de
años en el desarrollo humano. Como expresión coloquial el término childhood es sinónimo de infancia
en sentido amplio y cubriría desde el nacimiento hasta los 7-8 años e incluso
hasta el final de la adolescencia. Desde el punto de vista de la
psicología y sociología del desarrollo se suele dividir en early childhood (infancia temprana) que comenzaría cuando el niño
empieza a hablar o a andar (toddlerhood)
y continúa
aproximadamente hasta los 7 u 8 años. A partir de ese momento comenzaría
middle childhood (media infancia)
hasta el comienzo de la adolescencia
[40]
Gawlik A, Hochberg Z. (Op.cit)
[41] Hochberg Z: Evo Devo of Child Growth: Treatize on
Child Growth and Human Evolution New York: Wiley; 2012. (citado por Hochberg Z, Belsky J. Evo-devo of human
adolescence: beyond disease models of early puberty. BMC Medicine.
2013;11:113.
[42] Weisfeld GE, Czilli T, Phillips KA, Gall JA, Lichtman CM: Possible
olfaction-based mechanisms in human kin recognition and inbreeding avoidance. J
Exp Child Psychol. 2003, 85: 279-295
[43] Bogin B: Evolutionary
perspective on human growth. Annu Rev Anthropol 1999, 28:109-153.
[44] Tardieu C: Short
adolescence in early hominids: infantile and adolescent growth of the human
femur. Am J Phys Anthropol 1998, 107:163-178.
[45] Zemel BS, Katz
SH: The contribution of adrenal and gonadal androgens to the growth in height
of adolescent males. Am J Phys Anthropol 1986, 71:459-466.
[46] Hochberg Z: Juvenility in the context of life history
theory. Arch Dis Child 2008, 93:534-539.
[47] Palmert MR, Hayden DL, Mansfield MJ, Crigler JF Jr,
Crowley WF Jr, Chandler DW, Boepple PA: The longitudinal study of adrenal
maturation during gonadal suppression: evidence that adrenarche is a gradual
process. J Clin Endocrinol Metab 2001, 86:4536-4542.
[48] Arlt W, Martens JW, Song M, Wang JT, Auchus RJ,
Miller WL: Molecular evolution of adrenarche: structural and functional
analysis of p450c17 from four primate species. Endocrinology 2002,
143:4665-4672.
[49] Hochberg Z: Evo-devo of child growth II: human life
history and transition between its phases. Eur J Endocrinol 2009, 160:135-141.
[50] Suzuki M, Wright LS, Marwah P, Lardy HA, Svendsen CN:
Mitotic and neurogenic effects of dehydroepiandrosterone (DHEA) on human neural
stem cell cultures derived from the fetal cortex. Proc Natl Acad Sci USA 2004,
101:3202-3207.
[51] Hunt PJ, Gurnell EM, Huppert FA, Richards C, Prevost
AT, Wass JA, Herbert J, Chatterjee VK: Improvement in mood and fatigue after
dehydroepiandrosterone replacement in Addison’s disease in a randomized, double
blind trial. J Clin Endocrinol Metab
2000, 85:4650-4656
[52] Chen CC, Parker CR Jr: Adrenal androgens and the
immune system. Semin Reprod Med 2004, 22:369-377.
[53] Arquitt AB, Stoecker BJ, Hermann JS, Winterfeldt EA:
Dehydroepiandrosterone sulfate, cholesterol, hemoglobin, and anthropometric
measures related to growth in male adolescents. J Am Diet Assoc 1991,
91:575-579.
[54] Campbell B: Adrenarche and the evolution of human
life history. Am J Hum Biol 2006, 18(5):569-589.
[55] Hochberg Z,
Albertson-Wikland K. Evo-devo of infantile and childhood growth. Pediatr Res.
2008;64:2–7.
[56] Liu Y, Albertsson-Wikland K, Karlberg J. Long-term
consequences of early linear growth retardation (stunting) in Swedish children.
Pediatr Res. 2000;47:475–80
[57] Karlberg J, Jalil F, Lam B, Low L, Yeung CY. Linear
growth retardation in relation to the three phases of growth. Eur J Clin Nutr.
1994;48(suppl 1):25–43;discussion 43–4.
[58] Gawlik A, Walker RS, Hochberg Z. Impact of infancy
duration on adult size in 22 subsistence-based societies. Acta Paediatr 2011;100(12):248–52.
[59] Gawlik A, Walker RS, Hochberg Z. (Ibidem)
[60] Bogin B, Silva
MI, Rios L. Life history trade-offs in human growth: adaptation or pathology?
Am J Hum Biol. 2007;19:631–42.
[61] Hochberg Z, Gawlik A, Walker RS. Evolutionary fitness
as a function of pubertal age in 22 subsistence-based traditional societies.
Int J Pediatr Endocrinol. 2011;2011(1)2 Epub 2011 Jun 21.
[62] Schneider JE. Energy balance and reproduction.
Physiology and Behavior. 2004;81:289–317.
[63] Sisk CL, Foster DL. The neural basis of puberty and
adolescence. Nat Neurosci. 2004;7:1040–7.
[64] Templeton A: Has
human evolution stopped? Rambam
Maimonides Medical Journal 2010, 1:e0006:1-10
[65] Ellis BJ: Timing of pubertal maturation in girls: an
integrated life history approach. Psychol Bull 2004, 130(6):920-958.
[66] . Ong KK, Elks CE, Li S, Zhao JH, Luan J, Andersen
LB, Bingham SA, Brage S, Smith GD, Ekelund U, Gillson CJ, Glaser B, Golding J,
Hardy R, Khaw KT, Kuh D, Luben R, Marcus M, McGeehin MA, Ness AR, Northstone K,
Ring SM, Rubin C, Sims MA, Song K, Strachan DP, Vollenweider P, Waeber G,
Waterworth DM, Wong A, et al: Genetic variation in LIN28B is associated with the
timing of puberty. Nat Genet 2009, 41:729-733.
[67] Towne B, Czerwinski SA, Demerath EW, Blangero J,
Roche AF, Siervogel RM: Heritability of age at menarche in girls from the Fels
longitudinal study. Am J Phys Anthropol 2005, 128:210–219.
[68] Graber JA, Brooks- Gunn J, Warren MP: The antecedents
of menarcheal age: heredity, family environment, and stressful life events.
Child Dev 1995, 66:346–359.
[69] Kaprio J, Rimpela A, Winter T, Viken RJ, Rimpela M,
Rose RJ: Common genetic influences on BMI and age at menarche. Hum Biol 1995,
67:739–753
[70]
Silventoinen K, Haukka J, Dunkel L, Tynelius P, Rasmussen F. Genetics of
pubertal timing and its associations with relative weight in childhood and
adult height: the Swedish Young Male
Twins Study. Pediatrics. 2008 Apr;121(4):e885-91.
[71]
Towne B, Czerwinski SA, Demerath EW, Blangero J, Roche AF, Siervogel RM.
Heritability of age at menarche in girls from the Fels Longitudinal Study. Am J
Phys Anthropol. 2005;128:210–9.
[72]
De Vries L, Kauschansky A, Shohat M, Phillip M. Familial central precocious
puberty suggests autosomal dominant inheritance. J Clin Endocrinol Metab.
2004;89:1794–800.
[73]
Belsky J, 2012……VEr mas adealnte
[74]
Tena-Sempere M. Keeping puberty on time: novel signals and mechanisms involved.
Curr Top Dev Biol. 2013;105:299–329.
[75]
De Vries L, Kauschansky A, Shohat M, Phillip M. Familial central precocious
puberty suggests autosomal dominant inheritance. J Clin Endocrinol Metab.
2004;89:1794–800
[76]
Durand A, Bashamboo A, McElreavey K, Brauner R. Familial early puberty:
presentation and inheritance pattern in 139 families. BMC Endocr Disord. 2016
Sep 13;16(1):50.
[77] Grivas TB, Vasiliadis E, Mouzakis V, Mihas C,
Koufopoulos G: Association between adolescent idiopathic scoliosis prevalence
and age at menarche in different geographic latitudes. Scoliosis 2006, 23:1–9.
[78] Chumlea WC, Schubert CM, Roche AF, Kulin HE, Lee PA,
Himes JH, Sun SS: Age at menarche and racial comparisons in US girls.
Pediatrics 2003, 111:110–113.
[79] Frisch RE, Revelle R: Height and weight at menarche
and a hypothesis of critical body weights and adolescent events. Science 1970,
169:397–399.
[80] Esteva de Entonio, I; Soriguer
Escofet, F.J.C-Soriguer Escofet, R C; Martos, E; Esteban, M. Edad de la
menarquia y nutricion. Endocrinología. 1987, 34(1): 18-22, 3.
[81] Towne B, Czerwinski SA, Demerath EW, Blangero J,
Roche AF, Siervogel RM: Heritability of age at menarche in girls from the Fels
longitudinal study. Am J Phys Anthropol 2005, 128:210–219.
[82] Deardorff J1,
Ekwaru JP, Kushi LH, Ellis BJ, Greenspan LC, Mirabedi A, Landaverde EG, Hiatt
RA: Father absence, BMI, and pubertal timing in girls: differential effects by
family income and ethnicity. J Adolesc Health 2011, 48:441–447.
[83] Graber JA, Brooks- Gunn J, Warren MP: The antecedents
of menarcheal age: heredity, family environment, and stressful life events.
Child Dev 1995, 66:346–359.
[84] Gluckman PD, Hanson MA: Evolution, development and
timing of puberty. Trends Endocrinol Metab 2006, 17:7-12.
[85] Hochberg Z, Gawlik A, Walker RS: Evolutionary fitness
as a function of pubertal age in 22 subsistence-based traditional societies.
Int J Pediatr Endocrinol 2011, 2011:2.
[86] Cooper C, Kuh D, Egger P, Wadsworth M, Barker D:
Childhood growth and age at menarche. Br J Obstet Gynaecol 1996, 103:814-817.
[87] Ong KK, Potau N, Petry CJ, Jones R, Ness AR, Honour
JW, de Zegher F, Ibanez L, Dunger DB: Opposing influences of prenatal and
postnatal weight gain on adrenarche in normal boys and girls. J Clin Endocrinol
Metab 2004, 89:2647-2651
[88] Parent AS,
Teilmann G, Juul A, Skakkebaek NE, Toppari J, Bourguignon JP: The timing of
normal puberty and the age limits of sexual precocity: variations around the
world, secular trends, and changes after migration. Endocr Rev 2003,
24:668-693.
[89] Migliano AB,
Vinicius L, Lahr MM: Life history trade-offs explain the evolution of human
pygmies. Proc Natl Acad Sci USA 2007,
[90]
Euling SY, Op. cit.
[91]
Onland-Moret NC, Peeters PH, van Gils CH, Clavel-Chapelon F, Key T, Tjonneland
A, et al.: Age at menarche in relation to adult height: the EPIC study. Am J
Epidemiol 2005, 162:623-632.
[92]
Okasha M, McCarron P, McEwen J, Smith GD: Age at menarche: secular trends and
association with adult anthropometric measures. Ann Hum Biol 2001, 28:68-78
[93]
Clavel-Chapelon F, Gerber M: Reproductive factors and breast cancer risk. Do
they differ according to age at diagnosis? Breast Cancer Res Treat 2002,
72:107-11
[94]
Chodick G, Huerta M, Balicer RD, Davidovitch N, Grotto I: Secular trends in age
at menarche, smoking, and oral contraceptive use among Israeli girls. Prev
Chronic Dis 2005, 2:A12
[95]
Huen KF, Leung SS, Lau JT, Cheung AY, Leung NK, Chiu MC: Secular trend in the
sexual maturation of southern Chinese girls. Acta Paediatr 1997, 86:1121-1124.
[96]
Harris MA, Prior JC, Koehoorn M. Age at menarche in the Canadian population:
secular trends and relationship to adulthood BMI. J Adolesc Health. 2008
[97]
Nichols HB, Trentham-Dietz A, Hampton JM, Titus-Ernstoff L, Egan KM, Willett
WC, et al.: From menarche to menopause: trends among US Women born from 1912 to
1969. Am J Epidemiol 2006, 164:1003-1011.
[98]
Adanu RM, Hill AG, Seffah JD, Darko R, Anarfi JK, Duda RB. Secular trends in
menarcheal age among Ghanaian women in Accra. J Obstet Gynaecol. 2006
Aug;26(6):550-4.
[99] Wyshak G, Frish RE. Evidence for a secular trend in
the age of menarche. N Engl J Med. 1982;306(17):1033–1035
[100] Marrodan MD, Mesa MS,
Arechiga J, Perez-Magdaleno A: Trend in menarcheal age in Spain: rural and
urban comparison during a recent period. Ann Hum Biol 2000, 27:313-319.
[101] Euling SY, Op. cit.
[102]
Euling SY, Op. cit.
[103] Deng F, Tao FB, Liu DY, Xu YY, Hao JH, Sun Y, Su PY:
Effects of growth environments and two environmental endocrine disruptors on
children with idiopathic precocious puberty. Eur J Endocrinol 2012,
166:803-809.
1: Buck Louis GM, Gray LE Jr, Marcus M, Ojeda SR,
Pescovitz OH, Witchel SF, Sippell W, Abbott DH, Soto A, Tyl RW, Bourguignon JP,
Skakkebaek NE, Swan SH, Golub MS, Wabitsch M, Toppari J, Euling SY.
Environmental factors and puberty timing: expert panel research needs.
Pediatrics. 2008 Feb;121 Suppl 3:S192-207.
[105] Belsky J,
Steinberg L, Draper P: Childhood experience, interpersonal development, and
reproductive strategy: and evolutionary theory of socialization. Child Dev
1991, 62:647-670.
[106] Gluckman PD, Hanson MA: Evolution, development and
timing of puberty. Trends Endocrinol Metab 2006, 17:7-12.
[107] Hochberg Z, Belsky J. Evo-devo of human adolescence:
beyond disease models of early puberty. BMC Medicine. 2013;11:113.
doi:10.1186/1741-7015-11-113.
[108] Los huteritas son una
rama de los anabaptistas que,
como los amish y
los menonitas,
trazan sus raíces en la Reforma radical del siglo XVI.
Originarios de la provincia austríaca de Tirol en
el siglo XVI, los precursores de los
huteritas emigraron a Moravia para
escapar de la persecución. Allí, bajo la dirección de Jakob Hutter,
desarrollaron la forma comunal de la vida que incluye la propiedad comunitaria,
basada en los libros del Nuevo Testamento de los Hechos de los
Apóstoles
y en los Corintios 8:13-15. En 1873 emigraron a
Norteamérica asentándose en diferentes estados y en Canadá. Las tasas de
crecimiento de nacimiento son muy
elevadas, y aunque han descendido desde los años 1950 aun siguen siendo más del
doble de una población de referencia norteamericana (https://en.wikipedia.org/wiki/Hutterite)
Huterites
|
Dakota
del Sur
|
|
1950
|
45,9
|
23,4
|
1970
|
43.0
|
14.7
|
1990
|
35.2
|
12.1
|
[109] Pesonen AK, Raikkonen K, Heinonen K, Kajantie E,
Forsen T, Eriksson JG: Reproductive traits following a parent-child separation
trauma during childhood: a natural experiment during World War II. Am J Hum
Biol 2008, 20:345-351
[110] Belsky J, Steinberg L, Draper P. Childhood
experience, interpersonal development, and reproductive strategy: and
evolutionary theory of socialization. Child Dev. 1991 Aug;62(4):647-70.:
[111] Belsky J,
Steinberg L, Houts RM, Halpern-Felsher BL: The development of reproductive
strategy in females: early maternal harshness –> earlier menarche –>
increased sexual risk taking. Dev Psychol 2010, 46:120-128.
[112] Ellis BJ, Essex
MJ: Family environments, adrenarche, and sexual maturation: a longitudinal test
of a life history model. Child Dev 2007, 78:1799-1817.
[113] Belsky J, Houts RM, Fearon RM: Infant attachment
security and the timing of puberty: testing an evolutionary hypothesis. Psychol
Sci 2010, 21:1195-1201.
[114]
Belsky j The Development of Human Reproductive Strategies Progress and Prospects.
Current Direction in Psicological Science, 2012; 20 (5) 310-316.
[115] Cameron NM, Champagne FA, Parent C, Fish EW,
Ozaki-Kuroda K, Meaney MJ: The programming of individual differences in
defensive responses and reproductive strategies in the rat through variations
in maternal care. Neurosci Biobehav Rev 2005, 29:843-865.
[116] Manuck SB, Craig AE, Flory JD, Halder I, Ferrellc RE:
Reported early family environment covaries with menarcheal age as a function of
polymorphic variation in estrogen receptor-α (ESR). Dev Psychopathol 2011, 23:69-83.
[117] Janusz P, Kotwicka M,
Andrusiewicz M, Czaprowski D, Czubak J, Kotwicki T. Estrogen receptors genes
polymorphisms and age at menarche in idiopathic scoliosis. BMC Musculoskelet
Disord. 2014 Nov 19;15:383.
[118] Hochberg Z, Albertson-Wikland K. Evo-devo of
infantile and childhood growth. Pediatr Res. 2008;64:2–7.
[119] Heijmans BT, Tobi EW, Stein
AD, Putter H, Blauw GJ, Susser ES, Slagboom PE, Lumey LH. Persistent epigenetic
differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A.
2008 Nov 4;105(44):17046-9
[120] Tobi EW, Lumey LH, Talens
RP, Kremer D, Putter H, Stein AD, Slagboom PE, Heijmans BT. DNA methylation
differences after exposure to prenatal famine are common and timing- and sex-specific.
Hum Mol Genet. 2009
Nov 1;18(21):4046-53.
[122]
Coelho M, Oliveira T, Fernandes R. Biochemistry of adipose tissue: an endocrine
organ. Arch Med Sci. 2013 Apr 20;9(2):191-200.
[123] Poissonnet CM, Burdi AR, Bookstein FL. Growth and
development of human adipose tissue during early gestation. EarlyHum Dev
1983;8:1–11.
[124] Hauner H, Much D, Vollhardt C, Brunner S, Schmid D,
Sedlmeier E-M, Heimberg E, Schuster T, Zimmermann A, Schneider KTM, et al.
Effect of reducing the n26/n23 long-chain polyunsaturated fatty acid (LCPUFA)
ratio during pregnancy and lactation on infant adipose tissue growth within the
first year of life (INFAT-study): an open-label, randomized, controlled trial.
Am J Clin Nutr 2012;95:383–94.
[125] Veldhuis JD, Roemmich JN,
Richmond EJ, Rogol AD, Lovejoy JC, Sheffield-Moore M, Mauras N, Bowers CY.
Endocrine control of body composition in infancy, childhood, and puberty.
Endocr Rev. 2005 Feb;26(1):114-46.
[126] Rolland-Cachera MF, Deheeger
M, Avons P, Guilloud-Bataille M, Patois E, Sempé M. Tracking adiposity patterns
from 1 month to adulthood. Ann Hum Biol 1987; 14:
219–222.
[127] Soriguer Escofet FJ, Esteva
de Antonio I, Tinahones FJ, Pareja A. Adipose tissue fatty acids and size and
number of fat cells from birth to 9 years of age--a cross-sectional study in 96
boys. Metabolism.
1996 Nov;45(11):1395-401
[128]
Charles Darwin. El origen
de las especies. http://www.rebelion.org/docs/81666.pdf
[129]
Charles Darwin. La variación de animales y plantas bajo domesticación
(1ª edición: 1868), La Catarata, 2008.
[130] Villamor E, Jansen EC.
Nutritional Determinants of the Timing of Puberty. Annu Rev Public Health.
2016;37:33-46.
[131] Koprowski C, Ross RK, Mack WJ, Henderson BE,
Bernstein L. 1999. Diet, body size and menarche in a multiethnic cohort. Br.
J. Cancer 79:1907–11
[132]
Mesa JM,
Araujo C, Horta BL, Gigante DP. 2010. Growth
patterns in early childhood and the onset of menarche before age twelve. Rev. Saude
Publica 44:249–60
[133] Mesa JM, Araujo
C, Horta BL, Gigante DP. 2010. Growth patterns
in early childhood and the onset of menarche before age twelve. Rev. Saude
Publica 44:249–60
[134] Kuzawa CW, McDade TW, Adair LS, Lee N. 2010. Rapid
weight gain after birth predicts life history and reproductive strategy in
Filipino males. PNAS 107:16800–5
[135] Häger A,
Sjo¨stro¨m L, Arvidsson B, Bjo¨rntorp P, Smith U. Body fat and adipose tissue
cellularity in infants: a longitudinal study. Metabolism 1977;26:607–14
[136] Sjöstrom L, William Olsson T. Prospective studies on
adipose tissue development in man. Int J Obes 1981;5:597–604.
[137] Salans LB, Cushman SW, Weismann RE. Studies of human
adipose tissue: adipose cell size and number in nonobese and obese patients. J
Clin Invest 1973;52:929–41.
[138] Baum D, Beck RQ, Hammer LD, Brasel JA, Greenwood MR.
Adipose tissue thymidine kinase activity in man. Pediatr Res 1986;20:118–21..
[139]
Knittle JL, Timmers K, Ginsberg-Fellner F, Brown RE, Katz DP. The growth of
adipose tissue in children and adolescents: cross-sectional and longitudinal
studies of adipose cell number and size. J Clin Invest 1979;63:239–46
[140] Haro-Mora JJ, García-Escobar
E, Porras N, Alcázar D, Gaztambide J, Ruíz-Órpez A, García-Serrano S, Rubio-Martín E,
García-Fuentes E, López-Siguero JP, Soriguer F, Rojo-Martínez G. Children whose
diet contained olive oil had a lower likelihood of increasing their body mass
index Z-score over 1 year. Eur J Endocrinol. 2011 Sep;165(3):435-9.
[141]
Hauner H, Brunner S, Amann-Gassner U. The role of dietary fatty acids for early
human adipose tissue growth. Am
J Clin Nutr. 2013 Aug;98(2):549S-55S.
[142]
Esteva de
Entonio, I; Soriguer Escofet, F.J.C-Soriguer Escofet, R C; Martos, E; Esteban,
M. Edad de la menarquia y nutricion. Endocrinología. 1987, 34(1):
18-22, 3.
[143] Soriguer FJ, González-Romero S,
Esteva I, García-Arnés JA, Tinahones F, Ruiz de Adana MS, Olveira G, Mancha I,
Vazques F. Does the intake of nuts and seeds alter the appearance of menarche? Acta
Obstet Gynecol Scand. 1995 Jul;74(6):455-61
[144] F.J-C-Soriguer Escofet.
Nutricióny Maduración Sexual. Endocrinología 34(3) ; 52-57, 1987
[145] Kennedy GC. The
development with age of hypothalamic restraint upon the appetite of the rat. J
Endocrinol. 1957 Nov;16(1):9-17.
[146]
Kennedy GC, Mitra J. Body weight and food intake as initiating factors for
puberty in the rat. J Physiol. 1963 Apr;166:408-18
[147] Engelbregt MJ, Houdijk ME, Popp-Snijders C, Delemarre-van de
Waal HA. The effects of
intra-uterine growth retardation and postnatal undernutrition on onset of
puberty in male and female rats. Pediatr Res.2000;48(6) :803– 807
[148]
Tanner JM. Growth of the human at the time of adolescence. Lect Sci Basis Med.
1953;1:308-63
[149] Frisch RE, Revelle R. Height
and weight at menarche and a hypothesis of critical body weights and adolescent
events. Science.
1970 Jul 24;169(3943):397-9.
[150] Esteva de Entonio, I (Op.cit).
[152] Biro FM, McMahon RP, Striegel-Moore R,
Crawford PB, Obarzanek E, Morrison JA,
Barton BA, Falkner F. Impact of timing of pubertal maturation on growth
in black and white female adolescents:
The National Heart, Lung, and Blood Institute Growth and Health Study. J
Pediatr. 2001 May;138(5):636-43
[153] Kaplowitz PB, Slora EJ,
Wasserman RC, Pedlow SE, Herman-Giddens ME. Earlier onset of puberty in girls:
relation to increased body mass index and race.Pediatrics.2001;108(2) :347– 353
[154] Freedman
DS, Khan LK, Serdula MK, Dietz WH, Srinivasan SR, Berenson GS. Relation of
menarche to race, time period, and anthropomorphic dimensions: the Bogalusa
Heart Study. Pediatrics.2002;110(4) .
Available at:www.pediatrics.org/cgi/content/full/110/4/e43
[155] Himes JH, Obarzanek E,
Baranowski T, Wilson DM, Rochon J, McClanahan BS. Early sexual maturation, body
composition, and obesity in African-American girls. Obes Res.2004;12(suppl) :64S– 72S
[156] Anderson SE, Dallal GE, Must A.
Relative weight and race influence average age at menarche: results from two
nationally representative surveys of US girls 25 years apart. Pediatrics.2003;111(4 pt 1) :844– 850
[157] Wang Y. Is obesity associated
with earlier sexual maturation? A comparison of the association in American
boys vs girls. Pediatrics.2002;110(5) :903– 910
[158] Vizmanos B, Marti-Henneberg C.
Puberty begins with a characteristic subcutaneous body fat mass in each sex. Eur J Clin Nutr.2000;54(3) :203– 208
[159] Wang Y. 2002, (Op. Cit).
[160] Laron Z. Is obesity associated
with early sexual maturation?Pediatrics.2004;113(1 pt 1) :171– 172
[162] Rosenbaum M, Leibel RL. Role of
gonadal steroids in the sexual dimorphism in body composition and circulating
concentrations of leptin. J Clin Endocrinol Metab.1999;84(6) :1784– 1789
[163] He Q, Karlberg J. BMI gain in
childhood and its association with height gain, timing of puberty, and final
height. Pediatr Res.2001;49(2) :244– 251
[164]
Davison KK, Susman EJ, Birch LL. Percent body fat at age 5 predicts earlier
pubertal development among girls at age 9. Pediatrics. 2003 Apr;111(4 Pt
1):815-21.
[165]
Lee JM, Appugliese D, Kaciroti N, Corwyn RF, Bradley RH, Lumeng JC. Weight status
in young girls and the onset of puberty. Pediatrics. 2007 Mar;119(3):e624-30
[166] Sanchez-Garrido MA,
Tena-Sempere M. Metabolic control of puberty: roles of
leptin and kisspeptins. Horm Behav. 2013
Jul;64(2):187-94.
[167]
Kelsey MM, Zeitler PS. Insulin Resistance of Puberty. Curr Diab Rep. 2016
Jul;16(7):64.
[168] Buchan DS, Ollis S, Young JD, Thomas NE, Cooper SM,
et al. 2011. The effects of time and intensity of exercise on novel and
established markers of CVD in adolescent youth. Am. J. Hum. Biol. 23:517–26
[169]
Kaplowitz PB. Link between body fat and the timing of puberty. Pediatrics.2008
Feb;121 Suppl 3:S208-17
[170] Evans MC, Anderson GM. Neuroendocrine integration of nutritional
signals onreproduction. J Mol Endocrinol. 2017 Feb;58(2):R107-R128
[171] Williams NI, Berga SL & Cameron JL 2007
Synergism between psychosocial and metabolic stressors: impact on reproductive
function in cynomolgus monkeys. American
Journal of Physiology: Endocrinology and Metabolism 293 E270–E276
[172] Rogers NH. Brown adipose
tissue during puberty and with aging. Ann Med. 2015, Mar;47(2):142-9.
[173] V, ML, F, M, TA, HH. Changes
in brown adipose tissue in boys and girls during childhood and puberty. J
Pediatr. 2012;160:604–9 e1
[174] Gilsanz V, Hu HH, Kajimura
S. Relevance of brown adipose tissue in infancy and adolescence. Pediatr Res.
2013 Jan;73(1):3-9.
[175]
Cypess AM, Lehman S,Williams G, Tal I,Rodman D, GoldfineAB,
et al. Identification and importance of brown adipose tissue in adult
humans. N Engl J Med.2009;360:1509–17.
[176]
Chalfant JS, Smith ML,Hu HH, Dorey FJ,Goodarzian F, Fu CH,
et al. Inverse association between brown adipose tissue activation and
white adipose tissue accumulation in successfully treated pediatric malignancy.
Am J Clin Nutr.2012;95:1144–9
No hay comentarios:
Publicar un comentario